Reaksionet e komponimeve të koordinimit ndodhin gjithmonë në sferën e koordinimit të një metali me ligandët e lidhur në të. Prandaj, është e qartë se në mënyrë që të ndodhë diçka, ligandët duhet të jenë në gjendje të bien në këtë sferë. Kjo mund të ndodhë në dy mënyra:
- një kompleks koordinativ i pangopur lidh një ligand të ri
- në një sferë koordinimi tashmë të përfunduar, një ligand zëvendësohet nga një tjetër.
Ne jemi njohur tashmë me metodën e parë kur diskutuam për pangopjen e koordinimit dhe rregullin 18-elektronësh. Këtu do të merremi me të dytën.
Ligandët e çdo lloji mund të zëvendësohen në çdo kombinim
Por zakonisht funksionon rregull i pashprehur– numri i vendeve të zëna të koordinimit nuk ndryshon. Me fjalë të tjera, numri i elektroneve nuk ndryshon gjatë zëvendësimit. Zëvendësimi i një lloji ligandi me një tjetër është mjaft i mundshëm dhe shpesh ndodh në realitet. Le t'i kushtojmë vëmendje vetëm trajtimit të saktë të ngarkesave kur ndryshojmë ligand L në X-ligand dhe anasjelltas. Nëse e harrojmë këtë, atëherë gjendja e oksidimit të metalit do të ndryshojë, dhe zëvendësimi i ligandëve nuk është një proces i reduktimit të oksidimit (nëse gjeni ose gjeni një shembull të kundërt, më tregoni - do të kreditohet automatikisht si duhet larg, nëse nuk mund të vërtetoj se keni gabuar, dhe madje edhe në këtë rast, ju garantoj një kontribut pozitiv në karma).
Zëvendësimi që përfshin ligand hapto
Me ligandët më kompleksë nuk ka më vështirësi - thjesht duhet të mbani mend një rregull mjaft të qartë: numri i vendeve të ligandës (d.m.th., numri i përgjithshëm i ligandëve ose qendrave të ligandës X- ose L) ruhet. Kjo rrjedh drejtpërdrejt nga ruajtja e numërimit të elektroneve. Këtu janë shembuj të vetëkuptueshëm.

Le t'i kushtojmë vëmendje shembullit të fundit. Reagenti fillestar për këtë reaksion është dikloridi i hekurit FeCl 2 . Deri vonë, do të kishim thënë: "Është vetëm kripë, çfarë lidhje ka kimia e koordinimit me të?" Por ne nuk do t'ia lejojmë më vetes një injorancë të tillë. Në kiminë e metaleve në tranzicion nuk ka "vetëm kripëra"; çdo derivat janë komponime koordinuese, për të cilat zbatohen të gjitha konsideratat në lidhje me numërimin e elektroneve, konfigurimin d, ngopjen e koordinimit, etj. Dikloruri i hekurit, siç jemi mësuar ta shkruajmë, do të rezultonte një kompleks Fe(2+) i tipit MX 2 me konfigurim d 6 dhe numër elektronesh 10. Nuk mjafton! Mirë? Në fund të fundit, ne kemi kuptuar tashmë se ligandët mund të jenë të nënkuptuar. Për të bërë reaksionin na duhet një tretës dhe për reaksione të tilla ka shumë të ngjarë THF. Shpërbërja e një kripe kristalore hekuri në THF ndodh pikërisht sepse tretësi dhurues zë hapësira të lira dhe energjia e këtij procesi kompenson shkatërrimin. rrjetë kristali. Ne nuk do të ishim në gjendje ta shpërndajmë këtë "kripë" në një tretës që nuk ofron shërbime të tretjes së metaleve për shkak të bazës së Lewis. Në këtë rast, dhe në një milion të ngjashme, zgjidhja është thjesht një ndërveprim koordinues. Le të shkruajmë, vetëm për saktësi, rezultatin e tretësirës në formën e kompleksit FeX 2 L 4, në të cilin dy jone klori mbeten në sferën e koordinimit në formën e dy ligandëve X, megjithëse ka shumë të ngjarë që ato të zhvendosen gjithashtu nga molekulat e tretësit dhurues me formimi i një kompleksi të ngarkuar FeL 6 2+. Në këtë rast nuk është aq e rëndësishme. Sido që të jetë, ne mund të supozojmë me siguri se kemi një kompleks 18-elektronish si në të majtë ashtu edhe në të djathtë.
Zëvendësimi, shtimi dhe shpërbërja e ligandëve janë të lidhura ngushtë dhe pazgjidhshmërisht
Nëse kujtojmë kiminë organike, atëherë kishte dy mekanizma zëvendësimi në një atom karboni të ngopur - SN1 dhe SN2. Në të parën, zëvendësimi ndodhi në dy faza: zëvendësuesi i vjetër u largua fillimisht, duke lënë një orbital të zbrazët në atomin e karbonit, i cili më pas u pushtua nga një zëvendësues i ri me një palë elektrone. Mekanizmi i dytë supozonte se ikja dhe ardhja kryheshin njëkohësisht, në bashkëpunim, dhe procesi ishte njëfazor.
Në kiminë e komponimeve të koordinimit, është mjaft e mundur të imagjinohet diçka e ngjashme. Por shfaqet një mundësi e tretë, të cilën atomi i karbonit të ngopur nuk e kishte - së pari lidhim një ligand të ri, pastaj shkëputim të vjetrën. Menjëherë bëhet e qartë se ky opsion i tretë vështirë se është i mundur nëse kompleksi ka tashmë 18 elektrone dhe është i ngopur me koordinim. Por është mjaft e mundur nëse numri i elektroneve është 16 ose më pak, domethënë kompleksi është i pangopur. Le të kujtojmë menjëherë analogjinë e dukshme nga kimi organike– Zëvendësimi nukleofilik në një atom karboni të pangopur (në një unazë aromatike ose në një karbon karbonil) gjithashtu ndodh fillimisht si shtimi i një nukleofili të ri, dhe më pas eliminimi i atij të vjetër.
Pra, nëse kemi 18 elektrone, atëherë zëvendësimi ndodh si një shtesë-abstraksion (dashësit e fjalëve "të zgjuara" përdorin termin mekanizëm disociativ-asociativ ose thjesht disociues). Një mënyrë tjetër do të kërkonte zgjerimin e sferës së koordinimit në një numër prej 20 elektronesh. Kjo nuk është absolutisht e pamundur, dhe opsione të tilla ndonjëherë edhe konsiderohen, por është padyshim shumë joprofitabile dhe çdo herë në rast dyshimi për një rrugë të tillë, kërkohen prova shumë domethënëse. Në shumicën e këtyre historive, studiuesit përfundimisht arritën në përfundimin se ata kishin anashkaluar ose kishin humbur diçka, dhe mekanizmi shoqërues u refuzua. Pra, nëse kompleksi origjinal ka 18 elektrone, atëherë së pari duhet të largohet një ligand, pastaj një i ri duhet të zërë vendin e tij, për shembull:

Nëse duam të fusim një hapto-ligand që zë disa vende në sferën e koordinimit, atëherë së pari duhet t'i lirojmë të gjitha. Si rregull, kjo ndodh vetëm në kushte mjaft të rënda, për shembull, për të zëvendësuar tre karbonile në karbonil kromi me η6-benzen, përzierja nxehet nën presion për shumë orë, duke lëshuar monoksidin e karbonit të lëshuar herë pas here. Megjithëse diagrami përshkruan ndarjen e tre ligandëve me formimin e një kompleksi shumë të pangopur me 12 elektrone, në realitet reagimi ka shumë të ngjarë të ndodhë në faza, duke lënë një karbonil në të njëjtën kohë dhe benzeni hyn në sferë, duke rritur gradualisht hapticitetin, përmes fazat minus CO - digapto - minus një CO më shumë - tetrahapto - minus një CO më shumë - heksagapto, në mënyrë që të mos fitohen më pak se 16 elektrone.

Pra, nëse kemi një kompleks me 16 elektrone ose më pak, atëherë zëvendësimi i ligandit ka shumë të ngjarë si një shtesë-eliminim (për ata që pëlqejnë fjalët me tinguj të thellë: asociativ-dissociativ ose thjesht asociativ): ligandi i ri vjen së pari. , pastaj i vjetri largohet. Ngrihen dy pyetje të dukshme: pse largohet ligandi i vjetër, sepse 18 elektrone janë shumë të mira dhe pse të mos bëjmë të kundërtën në këtë rast, si në komplekset me 18 elektrone. Pyetja e parë është e lehtë për t'u përgjigjur: çdo metal ka zakonet e veta, dhe disa metale, veçanërisht ato të vonshme, me d-predha pothuajse plotësisht të mbushura, preferojnë numërimin 16 elektrone dhe llojet përkatëse strukturore, dhe për këtë arsye hedhin jashtë ligandën shtesë. , duke u kthyer në konfigurimin e tyre të preferuar. Ndonjëherë faktori hapësinor gjithashtu ndërhyn me çështjen; ligandët ekzistues janë të mëdhenj dhe ai shtesë ndihet si një pasagjer autobusi në orën e pikut. Është më e lehtë të zbresësh dhe të ecësh sesa të vuash kështu. Sidoqoftë, mund të shtyni një pasagjer tjetër, ta lini të bëjë një shëtitje dhe ne do të shkojmë. Pyetja e dytë është gjithashtu e thjeshtë - në këtë rast, mekanizmi disociues së pari duhet të japë një kompleks 14-elektronish, dhe kjo është rrallë e dobishme.
Ja një shembull. Për shumëllojshmëri, le të zëvendësojmë X-ligand me një L-ligand dhe nuk do të ngatërrohemi për gjendjet dhe ngarkesat e oksidimit. Edhe një herë: pas zëvendësimit, gjendja e oksidimit nuk ndryshon, dhe nëse ligandi X është larguar, atëherë humbja duhet të kompensohet nga ngarkesa në metal. Nëse e harrojmë këtë, atëherë numri i oksidimit do të zvogëlohej me 1, por kjo është e pasaktë.

Dhe një gjë tjetër e çuditshme. Një lidhje metal-piridine u formua për shkak të çiftit të vetëm në azot. Në kiminë organike, në këtë rast do të tregonim patjetër një plus në azotin e piridinës (për shembull, me protonimin ose formimin e një kripe kuaternare), por ne kurrë nuk e bëjmë këtë në kiminë e koordinimit as me piridinën, as me ndonjë L-ligand tjetër. Kjo është jashtëzakonisht e bezdisshme për të gjithë ata që janë mësuar me sistemin e rreptë dhe të paqartë të vizatimit të strukturave në kiminë organike, por do të duhet të mësoheni me të, nuk është aq e vështirë.
Por nuk ka një analog të saktë të SN2 në kiminë e komponimeve të koordinimit; ekziston një i largët, por është relativisht i rrallë dhe ne nuk kemi vërtet nevojë për të.
Ligandë të qëndrueshëm dhe labile
Nuk mund të flasim fare për mekanizmat e zëvendësimit të ligandit nëse jo për një rrethanë jashtëzakonisht të rëndësishme që do ta përdorim shumë: zëvendësimi i ligandit, qoftë asociativ apo disociativ, presupozon domosdoshmërisht shkëputjen e ligandit të vjetër. Dhe është shumë e rëndësishme për ne të dimë se cilët ligandë largohen lehtë dhe cilët largohen keq, duke preferuar të qëndrojnë në sferën e koordinimit të metalit.
Siç do ta shohim së shpejti, në çdo reagim disa nga ligandët mbeten në sferën e koordinimit dhe nuk ndryshojnë. Ligandë të tillë zakonisht quhen ligandë spektatorë (nëse nuk dëshironi fjalë kaq të thjeshta, "joshkencore", përdorni fjalë angleze spektator në transkriptim lokal, spektator, ligand-spektator, por, të lutem, jo spektator - kjo është e padurueshme!). Dhe disa marrin pjesë drejtpërdrejt në reagim, duke u shndërruar në produkte të reagimit. Ligandë të tillë quhen aktorë (jo aktorë!), domethënë aktivë. Është mjaft e qartë se aktorët ligandë duhet të futen dhe hiqen lehtësisht në sferën e koordinimit të metalit, përndryshe reagimi thjesht do të ngecë. Por është më mirë të lihen ligandët e spektatorëve në sferën e koordinimit për shumë arsye, por të paktën për një gjë kaq banale si nevoja për të shmangur zhurmën e panevojshme rreth metalit. Është më mirë që vetëm ligandët të jenë aktorë dhe sasitë e nevojshme mund të marrin pjesë në procesi i duhur. Nëse ka më shumë vende koordinimi të disponueshme sesa është e nevojshme, aktorë shtesë ligandë mund të ulen në to, madje edhe ata që do të marrin pjesë në reaksionet anësore, duke ulur rendimentin e produktit të synuar dhe selektivitetin. Për më tepër, ligandët e spektatorëve pothuajse gjithmonë kryejnë shumë funksione të rëndësishme, për shembull, ato sigurojnë tretshmërinë e komplekseve, stabilizojnë gjendjen e saktë të valencës së metalit, veçanërisht nëse nuk është mjaft e zakonshme, ndihmojnë fazat individuale, sigurojnë stereoselektivitet, etj. Nuk do ta deshifrojmë ende, sepse të gjitha këto do t'i diskutojmë në detaje kur të arrijmë te reagimet specifike.
Rezulton se disa nga ligandët në sferën e koordinimit duhet të jenë të lidhur fort dhe jo të prirur për shkëputje dhe zëvendësim nga ligandë të tjerë. Ligandë të tillë zakonisht quhen të qëndrueshme koordinativisht . Ose thjesht e qëndrueshme, nëse nga konteksti është e qartë se po flasim për forcën e lidhjeve të ligandëve, dhe jo për stabilitetin e tyre termodinamik, që nuk na shqetëson aspak.
Dhe quhen ligandët që hyjnë e dalin lehtësisht dhe me dëshirë dhe janë gjithmonë të gatshëm t'u lënë vendin të tjerëve labile koordinuese , ose thjesht labile, dhe këtu, për fat, nuk ka paqartësi.
Ciklobutadieni si ligand
Kjo është ndoshta më shembull i ndritshëm fakti që në sferën e koordinimit një molekulë shumë e paqëndrueshme mund të bëhet një ligand i shkëlqyer, dhe sipas definicionit, i qëndrueshëm koordinimi, vetëm sepse nëse guxon të lërë sferën e ngrohtë dhe komode jashtë, asgjë e mirë nuk e pret atë (çmimi i daljes do të jetë pikërisht energjia e destabilizimit antiaromatik).
Ciklobutadieni dhe derivatet e tij janë shembujt më të njohur të anti-aromaticitetit. Këto molekula ekzistojnë vetëm kur temperaturat e ulëta, dhe në një formë shumë të shtrembëruar - për t'u larguar sa më shumë nga antiaromatikiteti, cikli shtrembërohet në një drejtkëndësh të zgjatur, duke hequr delokalizimin dhe duke dobësuar maksimalisht konjugimin e lidhjeve të dyfishta (ky quhet ndryshe efekti Jahn-Teller i Lloji i dytë: një sistem i degjeneruar, dhe katrori i ciklobutadienit përfaqëson është një biradikal i degjeneruar, mbani mend rrethin Frost - ai shtrembërohet dhe zvogëlon simetrinë për të hequr degjenerimin).
Por në komplekse, ciklobutadieni dhe ciklobutadienet e zëvendësuara janë ligandë tetrahapto të shkëlqyer, dhe gjeometria e ligandëve të tillë është saktësisht një katror, me gjatësi të njëjta lidhjesh. Si dhe pse ndodh kjo është një histori më vete, dhe jo aq e qartë sa duket shpesh.
Ligandët labile koordinues
Duhet të kuptoni se nuk ka gardh prej betoni të armuar me tela me gjemba dhe kulla sigurie midis zonave të ligandëve labile dhe të qëndrueshme. Së pari, varet nga metali, dhe LMKO funksionon mirë në këtë kontekst. Për shembull, metalet e tranzicionit të vonë preferojnë ligandët e butë, ndërsa metalet e hershme të tranzicionit preferojnë ato të forta. Le të themi, jodidi mban shumë fort d 8 atomet e paladiumit ose platinit, por rrallë hyn fare në sferën e koordinimit të titanit ose zirkonit në konfigurimin d 0. Por në shumë komplekse metalike me veçori më pak të theksuara, jodidi manifestohet si një ligand plotësisht lakueshëm, duke u lënë lehtësisht të tjerëve.
Gjërat e tjera janë të barabarta:
- L-ligandët janë zakonisht më labile se ligandët X;
- qëndrueshmëria e ligandëve X përcaktohet nga ngurtësia/butësia dhe natyra e metalit;
- Ligandët "të nënkuptuar" janë shumë të lakueshëm: tretës dhe ura në dimerë dhe grupime, aq sa prania e tyre në sferën e koordinimit shpesh neglizhohet plotësisht dhe strukturat pa to vizatohen me një sferë koordinimi formalisht të pangopur;
- Ligandët dihapto, për shembull alkenet dhe alkinet, sillen si L-ligandë tipikë: ata janë zakonisht mjaft të pandryshueshëm;
- ligandët me hapticitet më të madh janë rrallë labile, por nëse një ligand polihapto mund të ndryshojë mënyrën e lidhjes në mono-hapto, ai bëhet më i paqëndrueshëm, për shembull, η3-alilet sillen në këtë mënyrë;
- Ligandet kelate që formojnë unaza kelate me 5 dhe 6 anëtarë janë të qëndrueshëm dhe kelate me më të vogla ose një numër i madh atomet e ciklit janë të paqëndrueshëm, të paktën në një qendër (unaza e kelatit hapet dhe ligandi mbetet i varur si i thjeshtë). Kështu sillet acetati, për shembull;
Ligandë të qëndrueshëm koordinativisht
Le t'i përsërisim të gjitha përsëri, vetëm nga ana tjetër
Në sferën e koordinimit të metaleve, në përgjithësi ruhen (koordinativisht të qëndrueshme):
- Kelatorë 5 dhe 6 anëtarësh;
- polihapto-ligandët: për të rrëzuar ciklopentadienilet ose benzenin (arenet) nga sfera e koordinimit, duhet të përdorni të gjitha llojet e teknikave speciale - ato thjesht nuk dalin, shpesh duke përballuar edhe ngrohjen e zgjatur;
- ligandët e lidhur me një metal me një proporcion të lartë të efektit π-dhurues (mbrapsht-dhurim);
- ligandë të butë për metale të tranzicionit të vonë;
- Ligandi "i fundit" në sferën e koordinimit.
Gjendja e fundit duket e çuditshme, por imagjinoni një kompleks që ka shumë ligandë të ndryshëm, ndër të cilët nuk ka absolutisht të qëndrueshëm (pa kelatorë ose polihapto-ligandë). Më pas, në reaksione, ligandët do të ndryshojnë, duke folur relativisht, në rendin e qëndrueshmërisë relative. Më pak labile dhe e fundit që mbetet. Ky truk ndodh, për shembull, kur përdorim komplekset e paladium fosfinës. Fosfinat janë ligandë relativisht të qëndrueshëm, por kur ka shumë prej tyre dhe metali është i pasur me elektrone (d 8, d 10), ato i lënë vendin, njëri pas tjetrit, ligandëve aktorë. Por ligandi i fundit i fosfinës zakonisht mbetet në sferën e koordinimit, dhe kjo është shumë e mirë nga pikëpamja e reaksioneve në të cilat marrin pjesë këto komplekse. Në këtë çështje të rëndësishme do të kthehemi më vonë. Këtu është një shembull mjaft tipik kur vetëm një fosfinë "e fundit" mbetet nga sfera fillestare e koordinimit të kompleksit të paladium fosfinës në reaksionin Heck. Ky shembull na afron shumë me konceptin më të rëndësishëm në reaksionet e komplekseve të metaleve në tranzicion - konceptin e kontrollit të ligandit. Do ta diskutojmë më vonë.

Rimetalizimi
Kur zëvendësoni disa ligandë me të tjerë, është e rëndësishme të mos e teproni me reaktivitetin e ligandit hyrës. Kur kemi të bëjmë me reaksione të molekulave organike, është e rëndësishme për ne që të dërgojmë saktësisht një molekulë të secilit reaktant në sferën e koordinimit. Nëse dy molekula hyjnë në vend të njërës, ekziston një probabilitet i lartë i reaksioneve anësore që përfshijnë dy ligandë identikë. Humbja e reaktivitetit është gjithashtu e mundur për shkak të ngopjes së sferës së koordinimit dhe pamundësisë për të futur në të ligandët e tjerë të nevojshëm për procesin e pritshëm. Ky problem veçanërisht shpesh lind kur nukleofile të forta anionike, për shembull, karbanionet, futen në sferën e koordinimit. Për të shmangur këtë, përdoren derivate më pak reaktivë, në të cilët, në vend të kationit të metalit alkal, i cili përcakton jonikitetin e lartë të lidhjes, përdoren më pak metale elektropozitive dhe metaloidë (zink, kallaj, bor, silikon etj.), duke formuar lidhjet kovalente me pjesën nukleofile . Reaksionet e derivateve të tillë me derivatet e metaleve kalimtare prodhojnë produkte të zëvendësimit të ligandit, në parim njëlloj sikur nukleofili të ishte në formë anionike, por për shkak të reduktimit të nukleofilicitetit me më pak komplikime dhe pa reaksione anësore.
Reaksione të tilla të zëvendësimit të ligandit zakonisht quhen transmetalacion për të theksuar faktin e qartë se nukleofili duket se ndryshon metalet - më elektropozitiv në më pak elektropozitiv. Prandaj, ky emër përmban një element të skizofrenisë së pakëndshme - dukej se tashmë kishim rënë dakord që do t'i shikonim të gjitha reagimet nga këndvështrimi i një metali kalimtar, por befas e humbëm përsëri dhe shikojmë këtë reagim dhe vetëm këtë reagim. nga pikëpamja e një nukleofili. Duhet të keni durim, kështu është zhvilluar dhe pranuar terminologjia. Në fakt, kjo fjalë kthehet në kiminë e hershme të përbërjeve organometalike dhe në faktin se veprimi i përbërjeve të litiumit ose organomagnezit në halogjenët e metaleve dhe metaloideve të ndryshme është një nga metodat kryesore për sintezën e të gjitha përbërjeve organometalike, kryesisht atyre në tranzicion. , dhe reagimi që ne po shqyrtojmë tani në kiminë e përbërjeve të koordinimit të metaleve në tranzicion - vetëm një përgjithësim metodë e vjetër kimi organometalike, nga e cila u rrit e gjitha.
![]()
Si ndodh transmetalizimi?
Rimetalizimi është i ngjashëm me zëvendësimin konvencional dhe jo i ngjashëm. Duket sikur - nëse e konsiderojmë një reagjent organometrik jo-kalimtar thjesht një karbanion me një kundërjon, atëherë lidhja karbon-metal jo-kalimtar është jonike. Por kjo ide duket të jetë e vërtetë vetëm për metalet më elektropozitive - magnezin. Por tashmë për zinkun dhe kallajin kjo ide është shumë larg së vërtetës.
Prandaj, dy lidhje σ dhe katër atome në skajet e tyre hyjnë në reaksion. Si rezultat, formohen dy lidhje të reja σ dhe katër atome lidhen me njëri-tjetrin në një rend të ndryshëm. Me shumë mundësi, e gjithë kjo ndodh njëkohësisht në një gjendje tranzicioni prej katër anëtarësh, dhe vetë reagimi ka një karakter të bashkërenduar, si shumë reagime të tjera të metaleve në tranzicion. Bollëku i elektroneve dhe orbitaleve për fjalë për fjalë për të gjitha shijet dhe për të gjitha llojet e simetrive i bën metalet në tranzicion të aftë për të mbajtur njëkohësisht lidhjet në gjendje tranzicioni me disa atome.
Në rastin e rimetalizimit, marrim një rast të veçantë të shumë procesi i përgjithshëm, e cila quhet thjesht metateza e lidhjes σ. Mos i ngatërroni vetëm me metatezën e vërtetë të olefinave dhe acetileneve, të cilat janë reaksione katalitike të plota me mekanizmat e tyre. Në këtë rast po flasim për mekanizmin e transmetalizimit ose një proces tjetër në të cilin ndodh diçka e ngjashme.

Hyrje në veprën
Rëndësia e punës. Komplekset e porfirinave me metale në gjendje të lartë oksidimi mund të koordinojnë bazat në mënyrë shumë më efikase sesa komplekset M 2+ dhe të formojnë komponime të përziera koordinuese në të cilat në sferën e parë të koordinimit të atomit të metalit qendror, së bashku me ligandin makrociklik, ka acidoligande jo-ciklike. dhe nganjëherë molekulat e koordinuara. Çështjet e përputhshmërisë së ligandit në komplekse të tilla janë jashtëzakonisht të rëndësishme, pasi porfirinat kryejnë funksionet e tyre biologjike në formën e komplekseve të përziera. Përveç kësaj, reaksionet e kthyeshme të shtimit (transferimit) të molekulave bazë, të karakterizuara nga konstante mesatarisht të larta të ekuilibrit, mund të përdoren me sukses për të ndarë përzierjet e izomerëve organikë, për analiza sasiore, për qëllime mjedisore dhe mjekësore. Prandaj, studimet e karakteristikave sasiore dhe stoikiometrisë së ekuilibrave të koordinimit shtesë mbi metaloporfirinat (MPs) dhe zëvendësimin e ligandëve të thjeshtë në to janë të dobishme jo vetëm nga pikëpamja njohuri teorike vetitë e metaloporfirinave si komponime komplekse, por edhe për zgjidhjen e problemit praktik të kërkimit të receptorëve dhe transportuesve të molekulave ose joneve të vogla. Deri më sot, studimet sistematike për komplekset e joneve metalike shumë të ngarkuara praktikisht mungojnë.
Qëllimi i punës. Punë e vërtetë i kushtohet studimit të reaksioneve të komplekseve të përziera që përmbajnë porfirinë të kationeve metalike shumë të ngarkuara Zr IV, Hf IV, Mo V dhe W V me baza N bioaktive: imidazol (Im), piridinë (Py), pirazinë (Pyz), benzimidazol. (BzIm), karakteristikat e stabilitetit dhe vetitë optike të komplekseve molekulare, vërtetimi i mekanizmave të reagimit hap pas hapi.
Risi shkencore. Duke përdorur metodat e titrimit spektrofotometrik të modifikuar, kinetikën kimike, thithjen elektronike dhe vibruese dhe spektroskopinë 1H NMR, për herë të parë u përftuan karakteristikat termodinamike dhe mekanizmat stoikiometrikë të reaksioneve të bazave N me metaloporfirina me një sferë koordinimi të përzier (X) n. -2 MTPP (X – acidoligand Cl - , OH) u vërtetuan - , O 2- , TPP - tetrafenilporfirin dianion). Është vërtetuar se në shumicën dërrmuese të rasteve, proceset e formimit të supramolekulave me bazë metaloporfirine zhvillohen hap pas hapi dhe përfshijnë disa reaksione elementare të kthyeshme dhe të ngadalta të pakthyeshme të koordinimit të molekulave bazë dhe zëvendësimit të ligandëve acidë. Për secilën fazë të reaksioneve hap pas hapi, stoikiometri, konstante të ekuilibrit ose shpejtësisë, u përcaktuan rendet e reaksioneve të ngadalta bazuar në bazën dhe produktet u karakterizuan në mënyrë spektrale (UV, spektrat e dukshëm për produktet e ndërmjetme dhe UV, të dukshme dhe IR për produktet përfundimtare). Për herë të parë janë marrë ekuacione korrelacioni që bëjnë të mundur parashikimin e qëndrueshmërisë së komplekseve supramolekulare me baza të tjera. Ekuacionet përdoren në punim për të diskutuar mekanizmin e detajuar të zëvendësimit të OH - në komplekset Mo dhe W nga një molekulë bazë. Janë përshkruar vetitë e MR, të cilat e bëjnë atë premtuese për përdorim në zbulimin, ndarjen dhe analizën sasiore të bazave biologjikisht aktive, të tilla si stabiliteti mesatarisht i lartë i komplekseve supramolekulare, një përgjigje optike e qartë dhe e shpejtë, një prag i ulët ndjeshmërie dhe një sekondë. koha e qarkullimit.
Rëndësia praktike e punës. Rezultatet sasiore dhe vërtetimi i mekanizmave stoikiometrikë të reaksioneve të formimit të komplekseve molekulare janë të një rëndësie të madhe për kiminë e koordinimit të ligandëve makreterociklik. Puna e disertacionit tregon se komplekset e përziera që përmbajnë porfirinë shfaqin ndjeshmëri dhe selektivitet të lartë ndaj bazave organike bioaktive, brenda pak sekondave ose minutave ato ofrojnë një përgjigje optike të përshtatshme për zbulimin praktik të reaksioneve me bazat - VOCs, përbërësit e barnave dhe produkteve ushqimore, për shkak të të cilat rekomandohen për përdorim si përbërës të sensorëve bazë në ekologji, Industria ushqimore, mjekësi dhe bujqësi.
Miratimi i punës. Rezultatet e punës u raportuan dhe u diskutuan në:
IX Konferenca ndërkombëtare mbi problemet e zgjidhjes dhe kompleksimit në zgjidhje, Ples, 2004; Simpoziumi XII mbi ndërveprimet ndërmolekulare dhe konformacionet e molekulave, Pushchino, 2004; Sesionet shkencore XXV, XXVI dhe XXIX të seminarit rus për kiminë e porfirinave dhe analogëve të tyre, Ivanovo, 2004 dhe 2006; VI Shkollë-konferencë e shkencëtarëve të rinj të vendeve të CIS mbi kiminë e porfirinave dhe komponimeve të ngjashme, Shën Petersburg, 2005; Shkolla e VIII Shkencore - Konferenca për Kimi Organike, Kazan, 2005; Konferenca shkencore gjithë-ruse "Përbërjet makrociklike natyrore dhe analogët e tyre sintetikë", Syktyvkar, 2007; Konferenca e XVI Ndërkombëtare për Termodinamikën Kimike në Rusi, Suzdal, 2007; Konferenca XXIII Ndërkombëtare e Chugaev për Kiminë e Koordinimit, Odessa, 2007; Konferenca Ndërkombëtare për Porfirinat dhe Ftalocianinat ISPP-5, 2008; Konferenca e 38-të Ndërkombëtare për Kiminë e Koordinimit, Izrael, 2008.
Reaksioni kryesor i zëvendësimit në tretësirat ujore, shkëmbimi i molekulave të ujit (22), është studiuar për një numër të madh të joneve metalike (Fig. 34). Shkëmbimi i molekulave të ujit në sferën e koordinimit të një joni metalik me pjesën më të madhe të molekulave të ujit të pranishme si tretës ndodh shumë shpejt për shumicën e metaleve, dhe për këtë arsye shpejtësia e një reagimi të tillë mund të studiohet kryesisht me metodën e relaksimit. Metoda përfshin prishjen e ekuilibrit të sistemit, për shembull nga një rritje e mprehtë e temperaturës. Në kushte të reja (më shumë temperaturë të lartë) sistemi nuk do të jetë më në ekuilibër. Më pas matet shkalla e ekuilibrit. Nëse mund të ndryshoni temperaturën e tretësirës brenda 10 -8 sek, atëherë mund të matni shpejtësinë e një reagimi që kërkon më shumë se një periudhë kohe për t'u përfunduar 10 -8 sek.
Është gjithashtu e mundur të matet shpejtësia e zëvendësimit të molekulave të koordinuara të ujit në jonet e ndryshme metalike me ligandët SO 2-4, S 2 O 3 2-, EDTA, etj. (26). Shpejtësia e këtij reagimi
varet nga përqendrimi i jonit të metalit të hidratuar dhe nuk varet nga përqendrimi i ligandit hyrës, gjë që bën të mundur përdorimin e ekuacionit të rendit të parë (27) për të përshkruar shpejtësinë e këtyre sistemeve. Në shumë raste, shpejtësia e reaksionit (27) për një jon metalik të caktuar nuk varet nga natyra e ligandit hyrës (L), qofshin molekula H 2 O ose SO 4 2-, S 2 O 3 2-, ose Jonet EDTA.
Ky vëzhgim, i shoqëruar me faktin se ekuacioni i shpejtësisë për këtë proces nuk përfshin përqendrimin e ligandit influencues, sugjeron që këto reaksione vazhdojnë me një mekanizëm në të cilin hapi i ngadaltë përfshin thyerjen e lidhjes midis jonit metalik dhe ujit. Përbërja që rezulton ka të ngjarë të koordinojë shpejt ligandët e afërt.
Insekt. 4 i këtij kapitulli u deklarua se jonet e metaleve të hidratuar më shumë të ngarkuar, si Al 3+ dhe Sc 3+, shkëmbejnë molekulat e ujit më ngadalë se jonet M2+ dhe M+; Kjo jep arsye për të supozuar se thyerja e lidhjeve luan një rol të rëndësishëm në fazën që përcakton shkallën e të gjithë procesit. Përfundimet e marra në këto studime nuk janë përfundimtare, por ato japin arsye për të besuar se proceset SN 1 janë të rëndësishme në reaksionet e zëvendësimit të joneve të metaleve të hidratuara.
Ndoshta komponimet komplekse më të studiuara janë amminat e kobaltit (III). Stabiliteti i tyre, lehtësia e përgatitjes dhe reagimet e ngadalta i bëjnë ato veçanërisht të përshtatshme për studime kinetike. Meqenëse studimet e këtyre komplekseve u kryen ekskluzivisht në tretësira ujore, fillimisht duhet të shqyrtojmë reagimet e këtyre komplekseve me molekulat e tretësit - ujin. U zbulua se në përgjithësi, molekulat e amoniakut ose aminës të koordinuara nga joni Co(III) zëvendësohen aq ngadalë nga molekulat e ujit sa që zakonisht konsiderohet zëvendësimi i ligandëve të tjerë përveç amineve.
Shpejtësia e reaksioneve të tipit (28) u studiua dhe u zbulua se ishte e rendit të parë në lidhje me kompleksin e kobaltit (X është një nga shumë anionet e mundshme).
Meqenëse në tretësirat ujore përqendrimi i H 2 O është gjithmonë afërsisht 55.5 M, atëherë është e pamundur të përcaktohet efekti i ndryshimit të përqendrimit të molekulave të ujit në shpejtësinë e reaksionit. Ekuacionet e shpejtësisë (29) dhe (30) për tretësirë ujore nuk janë të dallueshme eksperimentalisht, pasi k është thjesht e barabartë me k" = k". Prandaj, është e pamundur të thuhet nga ekuacioni i shpejtësisë së reagimit nëse H2O do të marrë pjesë në hapin e procesit përcaktues të shpejtësisë. Përgjigja në pyetjen nëse ky reaksion vazhdon nga mekanizmi S N 2 me zëvendësimin e jonit X me një molekulë H 2 O ose me mekanizmin S N 1 , i cili së pari përfshin disociimin e ndjekur nga shtimi i një molekule H 2 O, duhet të merret duke përdorur të dhëna të tjera eksperimentale.
Ky problem mund të zgjidhet me dy lloje eksperimentesh. Shkalla e hidrolizës (zëvendësimi i një joni Cl- për molekulë uji) ekstazë- + është afërsisht 10 3 herë më e lartë se shkalla e hidrolizës 2+. Rritja e ngarkesës së kompleksit çon në forcimin e lidhjeve metal-ligand dhe, rrjedhimisht, në frenimin e ndarjes së këtyre lidhjeve. Duhet të merret parasysh edhe tërheqja e ligandëve hyrës dhe lehtësimi i reagimit të zëvendësimit. Meqenëse u konstatua një rënie në normë ndërsa ngarkesa e kompleksit rritej, në këtë rast një proces disociativ (S N 1) duket më i mundshëm.
Një metodë tjetër provimi bazohet në studimin e hidrolizës së një sërë kompleksesh të ngjashme ekstazë- + . Në këto komplekse, molekula e etilendiaminës zëvendësohet nga diamina të ngjashme, në të cilat atomet e hidrogjenit në atomin e karbonit zëvendësohen nga grupet CH 3. Komplekset që përmbajnë diamina të zëvendësuara reagojnë më shpejt se kompleksi i etilendiaminës. Zëvendësimi i atomeve të hidrogjenit me grupet CH 3 rrit vëllimin e ligandit, duke e bërë më të vështirë që atomi metalik të sulmohet nga një ligand tjetër. Këto pengesa sterike ngadalësojnë reaksionin nëpërmjet mekanizmit S N 2. Prania e ligandëve të rëndë pranë atomit të metalit nxit procesin e disociimit, pasi heqja e njërit prej ligandëve redukton akumulimin e tyre në atomin e metalit. Rritja e vërejtur në shkallën e hidrolizës së komplekseve me ligandë të rëndë është dëshmi e mirë se reaksioni vazhdon sipas mekanizmit S N 1.
Pra, si rezultat i studimeve të shumta të komplekseve acidoamine Co(II), rezultoi se zëvendësimi i grupeve acido me molekula uji është një proces disociues në natyrë. Lidhja atom kobalt-ligand shtrihet në një vlerë të caktuar kritike përpara se molekulat e ujit të fillojnë të hyjnë në kompleks. Në komplekset me ngarkesë 2+ dhe më të lartë, prishja e lidhjes kobalt-ligand është shumë e vështirë dhe hyrja e molekulave të ujit fillon të luajë një rol më të rëndësishëm.
U zbulua se zëvendësimi i grupit acid (X -) në kompleksin e kobaltit (III) me një grup të ndryshëm nga molekula H2O, (31) së pari kalon përmes zëvendësimit të tij me një molekulë.
tretës - ujë, i ndjekur nga zëvendësimi i tij me grup i ri Y(32).
Kështu, në shumë reaksione me komplekset e kobaltit (III), shpejtësia e reaksionit (31) është e barabartë me shpejtësinë e hidrolizës (28). Vetëm joni hidroksil ndryshon nga reagentët e tjerë në reaktivitetin e tij me amminat Co(III). Ai reagon shumë shpejt me komplekset amine të kobaltit (III) (rreth 10 6 herë më shpejt se uji) sipas llojit të reagimit hidroliza bazë (33).
Ky reagim u zbulua të jetë i rendit të parë në lidhje me ligand zëvendësues OH - (34). Rendi i dytë i përgjithshëm i reaksionit dhe përparimi jashtëzakonisht i shpejtë i reaksionit sugjerojnë se joni OH është një reagent nukleofilik jashtëzakonisht efektiv për komplekset Co(III) dhe se reaksioni vazhdon nëpërmjet mekanizmit SN2 nëpërmjet formimit të një ndërmjetësi.
Megjithatë, kjo veti e OH - mund të shpjegohet edhe me një mekanizëm tjetër [ekuacionet (35), (36)]. Në reagimin (35), kompleksi 2+ sillet si një acid (sipas Brønsted), duke dhënë kompleksin +, i cili është amido-(përmban)-komponim - bazë që i përgjigjet acidit 2+.
Më pas, reaksioni vazhdon nëpërmjet mekanizmit S N 1 (36) për të formuar një ndërmjetës me pesë koordinata, i cili më tej reagon me molekulat e tretësit për të prodhuar produktin përfundimtar të reaksionit (37). Ky mekanizëm reagimi është në përputhje me shpejtësinë e një reaksioni të rendit të dytë dhe korrespondon me mekanizmin S N 1. Meqenëse reaksioni në fazën e përcaktimit të shpejtësisë përfshin një konjugim bazë me kompleksin origjinal - acidin, këtij mekanizmi i jepet emërtimi S N. 1 CB.
Përcaktimi se cili prej këtyre mekanizmave shpjegon më mirë vëzhgimet eksperimentale është shumë i vështirë. Megjithatë, ka prova bindëse për të mbështetur hipotezën S N 1CB. Argumentet më të mira në favor të këtij mekanizmi janë si më poshtë: komplekset oktaedrale Co(III) përgjithësisht reagojnë nëpërmjet mekanizmit disociues S N 1 dhe nuk ka asnjë argument bindës pse joni OH- duhet të ndërmjetësojë procesin S N 2. Është vërtetuar se Joni hidroksil është një reagent i dobët nukleofilik në reaksionet me Pt(II), dhe për këtë arsye reaktiviteti i tij i pazakontë me Co(III) duket i paarsyeshëm. Reaksionet me komponimet e kobaltit (III) në mjedise jo ujore ofrojnë dëshmi të shkëlqyera për formimin e ndërmjetësve me pesë koordinata të ofruara nga mekanizmi S N 1 SV.
Prova përfundimtare është fakti se në mungesë të lidhjeve N - H në kompleksin Co(III), ai reagon ngadalë me jonet OH -. Kjo, natyrisht, sugjeron që vetitë acid-bazë të kompleksit janë më të rëndësishme se vetitë nukleofile të OH për shpejtësinë e reaksionit." Ky reagim i hidrolizës bazë të komplekseve të aminës Co(III) ilustron faktin se të dhënat kinetike shpesh mund të interpretohet në më shumë se një mënyrë, dhe për të përjashtuar një ose një tjetër mekanizëm të mundshëm, është e nevojshme të kryhet një eksperiment mjaft delikate.
Aktualisht, janë studiuar reaksionet e zëvendësimit të një numri të madh të komponimeve oktaedrale. Nëse marrim parasysh mekanizmat e tyre të reagimit, më i zakonshmi është procesi disociativ. Ky rezultat nuk është i papritur pasi gjashtë ligandë lënë pak hapësirë rreth atomit qendror që grupet e tjera t'i bashkohen atij. Ka vetëm disa shembuj ku është demonstruar shfaqja e një ndërmjetësi me shtatë koordinata ose është zbuluar ndikimi i një ligandi ndërhyrës. Prandaj, mekanizmi S N 2 nuk mund të refuzohet plotësisht si rrugën e mundshme reaksionet e zëvendësimit në komplekset oktaedrale.
Me kusht reaksionet kimike komplekset ndahen në ligandë shkëmbyes, redoks, izomerizim dhe të koordinuar.
Shpërndarja parësore e komplekseve në sferën e brendshme dhe të jashtme përcakton shfaqjen e reaksioneve të shkëmbimit të joneve të sferës së jashtme:
X m + mNaY = Y m + mNaX.
Përbërësit e sferës së brendshme të komplekseve gjithashtu mund të marrin pjesë në proceset metabolike që përfshijnë ligandët dhe agjentin kompleks. Për të karakterizuar reaksionet e zëvendësimit të ligandëve ose të jonit metalik qendror, përdorni emërtimet dhe terminologjinë e propozuar nga K. Ingold për reaksionet e përbërjeve organike (Fig. 42), nukleofile S N dhe elektrofile Zëvendësimet S E:
Z + Y = z +X S N
Z + M"= z + M S E .
Sipas mekanizmit të reaksionit të zëvendësimit, ato ndahen (Fig. 43) në asociative ( S N 1 dhe S E 1 ) dhe disociative ( S N 2 dhe S E 2 ), që ndryshon në gjendjen e tranzicionit me një numër koordinimi të rritur dhe të zvogëluar.
Klasifikimi i një mekanizmi reagimi si shoqërues ose disociativ është një detyrë e vështirë eksperimentalisht e arritshme për të identifikuar një ndërmjetës me një numër koordinimi të reduktuar ose të rritur. Në këtë drejtim, mekanizmi i reagimit shpesh gjykohet në bazë të të dhënave indirekte mbi efektin e përqendrimit të reagentëve në shpejtësinë e reaksionit, ndryshimet në strukturën gjeometrike të produktit të reaksionit, etj.
Për të karakterizuar shkallën e reaksioneve të zëvendësimit të ligandit të komplekseve, laureat i Nobelit 1983 G. Taube (Fig. 44) propozoi përdorimin e termave "labil" dhe "inert" në varësi të kohës së reaksionit të zëvendësimit të ligandit, më pak ose më shumë se 1 minutë. Termat labile ose inerte janë karakteristika të kinetikës së reaksioneve të zëvendësimit të ligandit dhe nuk duhet të ngatërrohen me karakteristikat termodinamike të stabilitetit ose paqëndrueshmërisë së komplekseve.
Qëndrueshmëria ose inertiteti i komplekseve varet nga natyra e jonit kompleks dhe ligandëve. Në përputhje me teorinë e fushës së ligandit:
1. Komplekset oktaedrale 3 d metale kalimtare me shpërndarje të valencës ( n -1) d elektrone për sigmë* (p.sh ) MO-të e lirimit janë të paqëndrueshme.
4- (t 2g 6 e g 1) + H 2 O= 3- + CN - .
Për më tepër, sa më e ulët të jetë energjia e stabilizimit nga fusha kristalore e kompleksit, aq më e madhe është qëndrueshmëria e tij.
2. Komplekset oktaedrale 3 d metale kalimtare me sigma të lirë* lirim p.sh orbitalet dhe një shpërndarje uniforme e valencës ( n -1) d elektronet në orbitalet t 2 g (t 2 g 3, t 2 g 6) janë inerte.
[Co III (CN) 6] 3- (t 2 g 6 e g 0) + H 2 O =
[Cr III (CN) 6] 3- (t 2 g 3 e g 0) + H 2 O =
3. Plano-katror dhe oktaedral 4 d dhe 5 d metale në tranzicion që nuk kanë elektrone për sigma* MO-të e lirimit janë inerte.
2+ + H 2 O =
2+ + H 2 O =
Ndikimi i natyrës së ligandëve në shpejtësinë e reaksioneve të zëvendësimit të ligandëve konsiderohet në kuadrin e modelit të "ndikimit të ndërsjellë të ligandëve". Një rast i veçantë i modelit të ndikimit të ndërsjellë të ligandëve është ai i formuluar në vitin 1926 nga I.I. Koncepti i Chernyaev për ndikimin trans (Fig. 45) - "Labiliteti i ligandit në kompleks varet nga natyra e ligandit trans-lokalizuar" - dhe propozon një numër trans-ndikimesh të ligandëve: CO, CN -, C 2 H 4 > PR 3, H - > CH 3 -, SC (NH 2) 2 > C 6 H 5 -, NO 2 -, I -, SCN - > Br -, Cl - > py , NH 3 , OH - , H 2 O .
Koncepti i ndikimit trans na lejoi të justifikonim rregullat kryesore:
1. Rregulli i Peyrone- për shkak të veprimit të amoniakut ose amineve në tetrakloroplatinat ( II ) kaliumi merret gjithmonë me konfiguracion cis diklorodiamineplatin:
2 - + 2NH 3 = cis - + 2Cl - .
Meqenëse reaksioni vazhdon në dy faza dhe ligandi i klorurit ka një ndikim të madh trans, zëvendësimi i ligandit të dytë klorur me amoniak ndodh me formimin e cis-[ Pt (NH 3 ) 2 Cl 2 ]:
2- + NH 3 = -
NH 3 = cis -.
2. Rregulli i Jergensen - me veprimin e acidit klorhidrik në klorur tetramine platini ( II ) ose komponime të ngjashme përftohet në konfigurimin trans dichlorodi-ammineplatinum:
[ Pt (NH 3 ) 4 ] 2+ + 2 HCl = trans-[ Pt (NH 3 ) 2 Cl 2 ] + 2 NH 4 Cl .
Në përputhje me serinë e trans-ndikimeve të ligandëve, zëvendësimi i molekulës së dytë të amoniakut nga një ligand klorur çon në formimin e trans-[ Pt (NH 3 ) 2 Cl 2 ].
3. Reagimi i tiuresë i Kurnakov - produkte të ndryshme të reaksionit të tiuresë me izomerët gjeometrikë të trans-[ Pt (NH 3 ) 2 Cl 2 ] dhe cis- [ Pt ( NH 3 ) 2 Cl 2 ]:
cis - + 4Tio = 2+ + 2Cl - + 2NH 3 .
Karakter të ndryshëm produktet e reaksionit shoqërohen me ndikimin e lartë trans të tiuresë. Faza e parë e reaksioneve është zëvendësimi i ligandëve të klorurit tioure me formimin e trans- dhe cis-[ Pt (NH 3 ) 2 (Tio ) 2 ] 2+ :
trans-[Pt (NH 3) 2 Cl 2 ] + 2 Thio = trans-[ Pt (NH 3) 2 (Tio) 2 ] 2+
cis - + 2Thio = cis - 2+.
Në cis-[Pt (NH 3) 2 (Tio ) 2 ] 2+ dy molekula amoniaku në pozicion trans ndaj tiuresë i nënshtrohen zëvendësimit të mëtejshëm, gjë që çon në formimin 2+ :
cis - 2+ + 2Thio = 2+ + 2NH 3 .
Në trans-[Pt (NH 3) 2 (Tio ) 2 ] 2+ dy molekula amoniaku me pak ndikim trans ndodhen në një pozicion trans me njëra-tjetrën dhe për këtë arsye nuk zëvendësohen nga tiurea.
Modelet e ndikimit trans u zbuluan nga I.I. Chernyaev kur studion reaksionet e zëvendësimit të ligandit në komplekset e platinit katror-planar ( II ). Më pas, u tregua se trans-ndikimi i ligandëve manifestohet edhe në komplekset e metaleve të tjera ( Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III )) dhe strukturë tjetër gjeometrike. Vërtetë, seritë e trans-ndikimit të ligandëve për metale të ndryshme janë disi të ndryshme.
Duhet të theksohet se ndikimi trans është efekti kinetik- sa më i madh të jetë ndikimi trans i një ligandi të caktuar, aq më shpejt ai zëvendësohet nga një ligand tjetër që është në një pozicion trans në lidhje me të.
Së bashku me efektin kinetik të ndikimit trans, në mes XX shekulli A.A. Grinberg dhe Yu.N. Kukushkin vendosi varësinë e trans-ndikimit të ligandit L nga ligandi i vendosur në pozicion cis në L . Kështu, studimi i shkallës së reaksionit të zëvendësimit Cl- amoniaku në komplekset e platinit ( II):
[PtCl 4 ] 2- + NH 3 = [ PtNH 3 Cl 3 ] - + Cl - K = 0,42. 10 4 l/mol. Me
[ PtNH 3 Cl 3 ] - + NH 3 = cis- [ Pt (NH 3 ) 2 Cl 2 ] + Cl - K = 1,14. 10 4 l/mol. Me
trans-[ Pt (NH 3) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2,90 . 10 4 l/mol. Me
tregoi se prania e një ose dy molekulave të amoniakut në pozicionin cis ndaj ligandit të klorurit të zëvendësuar çon në një rritje të qëndrueshme të shpejtësisë së reagimit. Ky efekt kinetik quhet ndikimi cis. Aktualisht, të dy efektet kinetike të ndikimit të natyrës së ligandëve në shpejtësinë e reaksioneve të zëvendësimit të ligandit (efekti trans- dhe cis-efekti) janë të kombinuara në një koncept të përgjithshëm. ndikimi i ndërsjellë i ligandëve.
Argumentimi teorik i efektit të ndikimit të ndërsjellë të ligandëve është i lidhur ngushtë me zhvillimin e ideve për lidhjet kimike në përbërjet komplekse. Në vitet '30 XX shekulli A.A. Greenberg dhe B.V. Nekrasov e konsideroi ndikimin trans brenda kornizës së modelit të polarizimit:
1. Efekti trans është tipik për komplekset, joni metalik qendror i të cilave është shumë i polarizueshëm.
2. Aktiviteti trans i ligandëve përcaktohet nga energjia e polarizimit të ndërsjellë të ligandit dhe jonit metalik. Për një jon metalik të caktuar, ndikimi trans i ligandit përcaktohet nga polarizimi i tij dhe distanca nga joni qendror.
Modeli i polarizimit është në përputhje me të dhënat eksperimentale për komplekset me ligandë të thjeshtë anionikë, të tillë si jonet halide.
Në vitin 1943 A.A. Greenberg hipotezoi se aktiviteti trans i ligandëve lidhet me vetitë e tyre reduktuese. Zhvendosja e densitetit të elektroneve nga ligandi i trans-lokalizuar në metal redukton ngarkesën efektive të jonit metalik, gjë që çon në një dobësim të lidhjes kimike me ligandin e vendosur trans.
Zhvillimi i ideve rreth ndikimit trans është i lidhur me aktivitetin e lartë trans të ligandëve bazuar në molekulat organike të pangopura si etileni në [ Pt(C2H4)Cl3 ] -. Sipas Chatt dhe Orgel (Fig. 46), kjo është për shkak tëpi-ndërveprimi dhan i ligandëve të tillë me metalin dhe mekanizmi shoqërues i reaksioneve të zëvendësimit për ligandët e vendosur trans. Koordinimi me jonin metalik të ligandit sulmues Z çon në formimin e një ndërmjetësi bipiramidal trigonal me pesë koordinata, i ndjekur nga eliminimi i shpejtë i ligandit lënë X. Formimi i një ndërmjetësi të tillë lehtësohet ngapi-bashkëveprim dative ligand-ligand metal Y , i cili redukton densitetin elektronik të metalit dhe zvogëlon energjinë e aktivizimit të gjendjes së tranzicionit me zëvendësimin e shpejtë të mëvonshëm të ligandit X.
Së bashku me fq pranuesi (C 2 H 4 , CN - , CO ...) ligandët që formojnë një ligand metalik dhanor lidhje kimike, kanë një ndikim të lartë trans dhesLigandët e donatorëve: H - , CH 3 - , C 2 H 5 - ... Trans-ndikimi i ligandëve të tillë përcaktohet nga ndërveprimi dhurues-pranues i ligandit X me metalin, i cili ul densitetin e elektroneve të tij dhe dobëson lidhjen e metalit me ligandin që largohet. Y.
Kështu, pozicioni i ligandëve në serinë e trans-aktivitetit përcaktohet nga veprimi i kombinuar i sigma- donator dhe pi-vetitë e ligandëve - sigma- donator dhe pi-vetitë pranuese të ligandit rrisin trans-influencën e tij, ndërsapi-dobësohen donatorët. Cili nga këta përbërës të bashkëveprimit ligand-metal mbizotëron në efektin trans, gjykohet në bazë të llogaritjeve kimike kuantike të strukturës elektronike të gjendjes së tranzicionit të reaksionit.
Nje nga fazat kritike në katalizën e kompleksit metalik, ndërveprimi i substratit Y me kompleksin ndodh përmes tre mekanizmave:
a) Zëvendësimi i ligandit me një tretës. Kjo fazë zakonisht përshkruhet si shpërbërja e kompleksit
Thelbi i procesit në shumicën e rasteve është zëvendësimi i ligandit me një tretës S, i cili më pas zëvendësohet lehtësisht nga një molekulë e substratit Y.
b) Lidhja e një ligandi të ri në një koordinatë të lirë me formimin e një shoqërie të ndjekur nga shkëputja e ligandit të zëvendësuar
c) Zëvendësim sinkron (tipi S N 2) pa formim të ndërmjetëm
Në rastin e komplekseve Pt(II), shpejtësia e reaksionit përshkruhet shumë shpesh nga ekuacioni me dy rrugë
Ku k S Dhe k Y janë konstantet e shpejtësisë së proceseve që ndodhin në reaksionet (5) (me një tretës) dhe (6) me ligand Y. Për shembull,

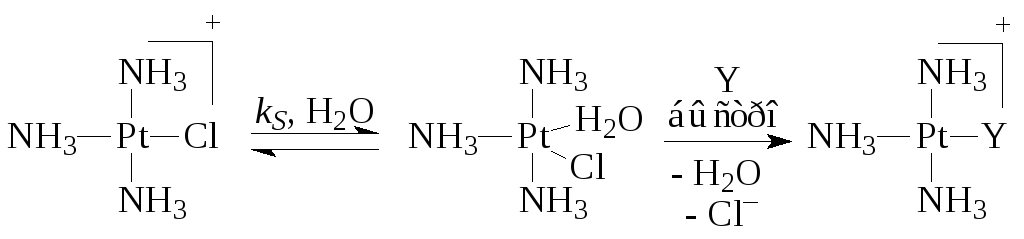
Faza e fundit e rrugës së dytë është shuma e tre fazave të shpejta elementare - eliminimi i Cl -, shtimi i Y dhe eliminimi i molekulës H 2 O.
Në komplekset katrore të sheshta të metaleve në tranzicion, vërehet një efekt trans, i formuluar nga I.I. Chernyaev - ndikimi i LT në shkallën e zëvendësimit të një ligandi të vendosur në një pozicion trans me ligand LT. Për komplekset Pt(II), efekti trans rritet në serinë e ligandëve:
H2O~NH 3 Prania e trans-efektit kinetik dhe trans-ndikimit termodinamik shpjegon mundësinë e sintetizimit të komplekseve izomere inerte të Pt(NH 3) 2 Cl 2: Reaksionet e zëvendësimit elektrofilik (S E) të hidrogjenit me një metal në sferën e koordinimit të metalit dhe proceset e tyre të anasjellta SH – H 2 O, ROH, RNH 2, RSH, ArH, RCCH. Edhe molekulat H 2 dhe CH 4 marrin pjesë në reaksione të këtij lloji Reaksionet e futjes së L përgjatë lidhjes M-X Në rastin e X=R (kompleks organometalik), molekulat e koordinuara me metal futen edhe në lidhjen M-R (L–CO, RNC, C 2 H 2, C 2 H 4, N 2, CO 2, O 2, etj. .). Reaksioni i futjes është rezultat i një sulmi intramolekular të një nukleofili në një molekulë të koordinuar ose . Reaksionet e kundërta – - dhe -reaksionet e eliminimit Reaksionet e shtimit oksidativ dhe eliminimit reduktiv M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4- Me sa duket, në këto reaksione ka gjithmonë një koordinim paraprak të molekulës së shtuar, por kjo nuk mund të zbulohet gjithmonë. Prandaj, prania e një zone të lirë në sferën e koordinimit ose e një vendi të lidhur me një tretës që zëvendësohet lehtësisht nga një substrat është një faktor i rëndësishëm që ndikon në reaktivitetin e komplekseve metalike. Për shembull, komplekset bis--alil të Ni janë prekursorë të mirë të specieve katalitikisht aktive, pasi për shkak të eliminimit të lehtë reduktiv të bis-alilit, shfaqet një kompleks me tretësin, i ashtuquajturi. nikel "i zhveshur". Roli i vendeve bosh ilustrohet nga shembulli i mëposhtëm: Reaksionet e shtimit nukleofilik dhe elektrofilik në komplekset dhe të metaleve Si ndërmjetës të reaksioneve katalitike, ekzistojnë të dyja përbërjet organometalike klasike që kanë lidhje M-C, M=C dhe MC, dhe komponime jo klasike në të cilat ligandi organik është i koordinuar sipas 2 , 3 , 4 , 5 dhe 6-lloji, ose është një element i strukturave me mungesë elektronesh - tejkalimi i grupeve CH 3 dhe C 6 H 6, karbide jo klasike (Rh 6 C(CO) 16, C(AuL) 5 +, C(AuL) 6 2+, etj.). Ndër mekanizmat specifikë për përbërjet -organometalike klasike, vëmë re disa mekanizma. Kështu, janë krijuar 5 mekanizma të zëvendësimit elektrofilik të atomit të metalit në lidhjen M-C. zëvendësimi elektrofilik me asistencën nukleofile AddEAdition-eliminim AdE(C) Shtimi në atomin C në hibridizimin sp 2 AdE(M) Shtesë oksiduese në metal Zëvendësimi nukleofilik në atomin e karbonit në reaksionet e demetalimit të përbërjeve organometalike ndodh si një proces redoks: Pjesëmarrja e mundshme e një agjenti oksidues në këtë fazë Një agjent i tillë oksidues mund të jetë CuCl 2, p-benzoquinone, NO 3 - dhe komponime të tjera. Këtu janë dy faza të tjera elementare karakteristike të RMX: hidrogjenoliza e lidhjes M-C dhe homoliza e lidhjes M-C Një rregull i rëndësishëm që zbatohet për të gjitha reaksionet e përbërjeve komplekse dhe organometalike dhe që lidhet me parimin e lëvizjes më të vogël është rregulli 16-18 i predhës elektronike të Tolman-it (Seksioni 2).

Reaksionet e ligandëve të koordinuar








Reaksionet e përbërjeve organometrike








