Koordinacinių junginių reakcijos visada vyksta metalo su jame susijungusiais ligandais koordinacinėje sferoje. Todėl akivaizdu, kad tam, kad kas nors išvis įvyktų, ligandai turi sugebėti patekti į šią sferą. Tai gali atsitikti dviem būdais:
- koordinatyviai nesotus kompleksas sujungia naują ligandą
- jau baigtoje koordinacinėje sferoje vienas ligandas pakeičiamas kitu.
Mes jau susipažinome su pirmuoju metodu, kai aptarėme koordinavimo neprisotinimą ir 18 elektronų taisyklę. Čia nagrinėsime antrąjį.
Bet kokio tipo ligandai gali būti pakeisti bet kokiu deriniu
Bet dažniausiai tai veikia neišsakyta taisyklė– užimtų koordinacinių vietų skaičius nesikeičia. Kitaip tariant, pakeitimo metu elektronų skaičius nesikeičia. Vieno tipo ligando pakeitimas kitu yra visiškai įmanomas ir dažnai pasitaiko realybėje. Atkreipkime dėmesį tik į teisingą įkrovų tvarkymą keičiant L ligandą į X ligandą ir atvirkščiai. Jei pamiršime apie tai, metalo oksidacijos būsena pasikeis, o ligandų keitimas nėra oksidacijos-redukcijos procesas (jei rasite ar sugalvosite priešingą pavyzdį, praneškite man - jis bus automatiškai įskaitytas teisingai toli, jei negaliu įrodyti, kad klydote, ir net šiuo atveju garantuoju teigiamą indėlį į karmą).
Pakeitimas naudojant hapto ligandus
Su sudėtingesniais ligandais daugiau sunkumų nekyla – tereikia atsiminti gana akivaizdžią taisyklę: ligandų vietų skaičius (tai yra bendras ligandų arba X arba L tipo ligandų centrų skaičius) išlaikomas. Tai tiesiogiai išplaukia iš elektronų skaičiavimo išsaugojimo. Čia yra savaime suprantamų pavyzdžių.

Atkreipkime dėmesį į paskutinį pavyzdį. Šios reakcijos pradinis reagentas yra geležies dichloridas FeCl 2 . Dar visai neseniai sakydavome: „Tai tik druska, ką su tuo turi koordinacinė chemija? Bet mes nebeleisime sau tokio nežinojimo. Pereinamųjų metalų chemijoje nėra „tiesiog druskų“, bet kokie dariniai yra koordinaciniai junginiai, kuriems galioja visi svarstymai apie elektronų skaičiavimą, d konfigūraciją, koordinacinį prisotinimą ir kt. Geležies dichloridas, kaip mes įpratę jį rašyti, būtų MX 2 tipo Fe(2+) kompleksas, kurio konfigūracija d 6 ir elektronų skaičius 10. Nepakanka! gerai? Juk mes jau išsiaiškinome, kad ligandai gali būti numanomi. Reakcijai atlikti mums reikia tirpiklio, o tokioms reakcijoms greičiausiai tai yra THF. Kristalinės geležies druskos ištirpimas THF vyksta būtent todėl, kad donorinis tirpiklis užima laisvą erdvę, o šio proceso energija kompensuoja sunaikinimą. kristalinė gardelė. Negalėtume šios „druskos“ ištirpinti tirpiklyje, kuris neteikia metalo tirpinimo paslaugų dėl Lewiso baziškumo. Šiuo atveju ir milijonu panašių sprendimų yra tiesiog koordinacinė sąveika. Tikslumo dėlei parašykime solvatacijos rezultatą FeX 2 L 4 komplekso pavidalu, kuriame koordinacinėje sferoje lieka du chloro jonai dviejų X ligandų pavidalu, nors greičiausiai juos taip pat išstumia donoro tirpiklio molekulių su įkrauto komplekso susidarymas FeL 6 2+. Šiuo atveju tai nėra taip svarbu. Bet kuriuo atveju galime drąsiai manyti, kad tiek kairėje, tiek dešinėje turime 18 elektronų kompleksą.
Ligandų pakeitimas, pridėjimas ir disociacija yra glaudžiai ir neatsiejamai susiję
Jei prisiminsime organinę chemiją, tai buvo du pakeitimo mechanizmai prie sočiojo anglies atomo - SN1 ir SN2. Pirmajame pakeitimas įvyko dviem etapais: senasis pakaitas pirmiausia paliko anglies atomo orbitalę, kurią vėliau užėmė naujas pakaitas su elektronų pora. Antrasis mechanizmas darė prielaidą, kad išėjimas ir atvykimas buvo vykdomi vienu metu, suderintai, o procesas buvo vieno etapo.
Koordinacinių junginių chemijoje visai įmanoma įsivaizduoti kažką panašaus. Bet atsiranda trečia galimybė, kurios sočiųjų anglies atomas neturėjo – iš pradžių prijungiame naują ligandą, tada atjungiame senąjį. Iš karto tampa aišku, kad šis trečiasis variantas vargu ar įmanomas, jei kompleksas jau turi 18 elektronų ir yra prisotintas koordinacijos. Bet tai visiškai įmanoma, jei elektronų skaičius yra 16 ar mažiau, tai yra, kompleksas yra nesotus. Iš karto prisiminkime akivaizdžią analogiją organinė chemija– nukleofilinis pakeitimas nesočiame anglies atome (aromatiniame žiede arba karbonilo anglies atome) taip pat pirmiausia įvyksta kaip naujo nukleofilo pridėjimas, o paskui senojo pašalinimas.
Taigi, jei turime 18 elektronų, tai pakeitimas įvyksta kaip abstrakcija-pridėjimas ("protingų" žodžių gerbėjai vartoja terminą disociatyvus-asociatyvus arba tiesiog disociacinis mechanizmas). Kitu būdu reikėtų išplėsti koordinavimo sferą iki 20 elektronų. Tai nėra visiškai neįmanoma, o tokie variantai kartais net svarstomi, tačiau tai tikrai labai nuostolinga ir kiekvieną kartą kilus įtarimui dėl tokio kelio reikalaujama labai reikšmingų įrodymų. Daugumoje šių istorijų tyrėjai galiausiai padarė išvadą, kad jie kažką nepastebėjo arba praleido, ir asociacinis mechanizmas buvo atmestas. Taigi, jei pradiniame komplekse yra 18 elektronų, pirmiausia turi išeiti vienas ligandas, tada jo vietą turi užimti naujas, pavyzdžiui:

Jei norime į koordinavimo sferą įtraukti kelias vietas užimantį hapto ligandą, pirmiausia turime juos visus atlaisvinti. Paprastai tai įvyksta tik esant gana atšiaurioms sąlygoms, pavyzdžiui, norint pakeisti tris karbonilus chromo karbonile η 6 -benzenu, mišinys kaitinamas slėgiu daug valandų, karts nuo karto išleidžiant išsiskiriantį anglies monoksidą. Nors diagramoje pavaizduota trijų ligandų disociacija, susidarant labai nesočiam kompleksui su 12 elektronų, iš tikrųjų reakcija greičiausiai vyksta etapais, paliekant po vieną karbonilą, o benzenas patenka į sferą, palaipsniui didindamas jautrumą. etapai minus CO - digapto - atėmus dar vieną CO - tetrahapto - atėmus dar vieną CO - heksagapto, kad nebūtų gauta mažiau nei 16 elektronų.

Taigi, jei turime kompleksą, kuriame yra 16 ar mažiau elektronų, tada ligando pakeitimas greičiausiai įvyksta kaip papildymas-eliminacija (tiems, kurie mėgsta giliai skambančius žodžius: asociatyvus-disociatyvus arba tiesiog asociatyvus): pirmiausia atsiranda naujas ligandas. , tada senasis palieka. Kyla du akivaizdūs klausimai: kodėl išeina senasis ligandas, nes 18 elektronų yra labai gerai, ir kodėl šiuo atveju nepadarius priešingai, kaip 18 elektronų kompleksuose. Į pirmąjį klausimą lengva atsakyti: kiekvienas metalas turi savo įpročius, o kai kurie metalai, ypač vėlyvieji, su beveik visiškai užpildytais d apvalkalais, teikia pirmenybę 16 elektronų skaičiui ir atitinkamiems struktūriniams tipams, todėl išmeta papildomą ligandą. , grįždami į mėgstamą konfigūraciją. Kartais reikalui trukdo ir erdvinis veiksnys, esami ligandai dideli, o papildomas jaučiasi kaip autobuso keleivis piko valandomis. Lengviau išlipti ir pasivaikščioti, nei šitaip kentėti. Tačiau galite išstumti kitą keleivį, leisti jam pasivaikščioti, ir mes eisime. Antrasis klausimas taip pat paprastas – tokiu atveju disociacinis mechanizmas pirmiausia turėtų duoti 14 elektronų kompleksą, o tai retai būna naudinga.
Štai pavyzdys. Dėl įvairovės pakeiskime X ligandą L ligandu ir nesusipainiosime dėl oksidacijos būsenų ir krūvių. Dar kartą: pakeitus, oksidacijos būsena nesikeičia, o jei X ligandas pasitraukė, nuostolius turi kompensuoti metalo krūvis. Jei apie tai pamirštume, oksidacijos skaičius sumažėtų 1, tačiau tai neteisinga.

Ir dar vienas keistas dalykas. Dėl vienišos azoto poros susidarė metalo-piridino jungtis. Organinėje chemijoje šiuo atveju neabejotinai parodytume piridino azoto pliusą (pavyzdžiui, protonuojant arba formuojant ketvirtinę druską), tačiau niekada to nedarome derindami chemiją su piridinu ar kitais L-ligandais. Tai baisiai erzina visus, kurie yra pripratę prie griežtos ir nedviprasmiškos organinės chemijos struktūrų braižymo sistemos, bet teks priprasti, tai nėra taip sunku.
Bet tikslaus SN2 analogo koordinacinių junginių chemijoje nėra, yra tolimas, bet jis yra gana retas ir mums jo tikrai nereikia.
Stabilūs ir labilūs ligandai
Apie ligandų pakeitimo mechanizmus išvis negalėtume kalbėti, jei ne viena itin svarbi aplinkybė, kurią naudosime daug: ligando pakeitimas, nesvarbu, asociatyvus ar disociatyvus, būtinai suponuoja senojo ligando disociaciją. Ir mums labai svarbu žinoti, kurie ligandai pasišalina lengvai, o kurie prastai, nori likti metalo koordinacinėje sferoje.
Kaip netrukus pamatysime, bet kurioje reakcijoje dalis ligandų lieka koordinacinėje sferoje ir nekinta. Tokie ligandai dažniausiai vadinami žiūrovų ligandais (jei nenorite tokių paprastų, „nemoksliškų“ žodžių, naudokite Angliškas žodisžiūrovas vietine transkripcija, žiūrovas, ligandas-žiūrovas, bet, prašau, ne žiūrovas – tai nepakeliama!). O kai kurie tiesiogiai dalyvauja reakcijoje, virsdami reakcijos produktais. Tokie ligandai vadinami veikėjais (ne veikėjais!), tai yra, aktyviaisiais. Visiškai aišku, kad ligandus-aktorius reikia lengvai įvesti ir pašalinti į metalo koordinacinę sferą, kitaip reakcija tiesiog užstrigs. Bet žiūrovų ligandus geriau palikti koordinacinėje sferoje dėl daugelio priežasčių, bet bent jau dėl tokios banalios, kaip poreikis vengti bereikalingo šurmulio aplink metalą. Geriau, kad tik ligandai būtų veikėjai ir reikiamus kiekius galėtų dalyvauti teisingas procesas. Jei laisvų koordinavimo vietų yra daugiau nei būtina, ant jų gali sėdėti papildomi ligandų veikėjai ir netgi tie, kurie dalyvaus šalutiniuose reakcijomis, sumažindami tikslinio produkto išeigą ir selektyvumą. Be to, žiūrovų ligandai beveik visada atlieka daug svarbių funkcijų, pavyzdžiui, užtikrina kompleksų tirpumą, stabilizuoja teisingą metalo valentinę būseną, ypač jei ji nėra visai įprasta, padeda atskiroms stadijoms, suteikia stereoselektyvumą ir kt. Kol kas neiššifruosime, nes visa tai išsamiai aptarsime, kai prieisime prie konkrečių reakcijų.
Pasirodo, kai kurie koordinacinės sferos ligandai turėtų būti tvirtai surišti ir nebūti linkę į disociaciją ir pakeitimą kitais ligandais. Tokie ligandai paprastai vadinami koordinaciniu požiūriu stabilus . Arba tiesiog stabilus, jei iš konteksto aišku, kad kalbame apie ligandų ryšių stiprumą, o ne apie jų pačių termodinaminį stabilumą, kuris mums visai nerūpi.
Ir vadinami ligandai, kurie lengvai ir noriai įeina ir išeina, ir visada pasiruošę užleisti vietą kitiems koordinacija labili , arba tiesiog labilus, ir čia, laimei, neaiškumų nėra.
Ciklobudienas kaip ligandas
Tai turbūt labiausiai ryškus pavyzdys faktas, kad koordinacinėje sferoje labai nestabili molekulė gali tapti puikiu ligandu ir pagal apibrėžimą koordinaciniu požiūriu stabili, jau vien todėl, kad jei išdrįs palikti šiltą ir jaukią sferą lauke, nieko gero jos nelauks (išėjimo kaina bus būtent tokia antiaromatinio destabilizavimo energija).
Ciklobutadienas ir jo dariniai yra geriausiai žinomi antiaromatiškumo pavyzdžiai. Šios molekulės egzistuoja tik tada, kai žemos temperatūros, ir labai iškreipta forma – siekiant kuo toliau nuo antiaromatiškumo, ciklas iškraipomas į pailgą stačiakampį, pašalinant delokalizaciją ir maksimaliai susilpninant dvigubų ryšių konjugaciją (tai kitaip vadinama Jahn-Teller efektu 2-oji rūšis: išsigimusi sistema, o ciklobutadieno kvadratas reiškia išsigimimą biradikalą, atsiminkite Frost apskritimą – jis yra iškraipytas ir sumažina simetriją, kad pašalintų degeneraciją).
Tačiau kompleksuose ciklobutadienas ir pakeisti ciklobutadienai yra puikūs tetrahapto ligandai, o tokių ligandų geometrija yra lygiai kvadratinė, su vienodais jungties ilgiais. Kaip ir kodėl tai atsitinka, yra atskira istorija ir nėra tokia akivaizdi, kaip dažnai manoma.
Koordinacijos labilūs ligandai
Reikia suprasti, kad tarp labilių ir stabilių ligandų zonų nėra gelžbetoninės tvoros su spygliuota viela ir apsauginiais bokšteliais. Pirma, tai priklauso nuo metalo, o LMKO puikiai veikia šiame kontekste. Pavyzdžiui, vėlyvieji pereinamieji metalai teikia pirmenybę minkštiesiems ligandams, o ankstyvieji pereinamojo laikotarpio metalai – kietiesiems. Tarkime, jodidas labai tvirtai laikosi paladžio ar platinos d 8 atomų, bet retai patenka į titano ar cirkonio koordinacinę sferą d 0 konfigūracijoje. Tačiau daugelyje metalų kompleksų, turinčių mažiau ryškių savybių, jodidas pasireiškia kaip visiškai labilus ligandas, lengvai užleidžiantis vietą kitiems.
Kiti dalykai yra vienodi:
- L ligandai paprastai yra labilesni nei X ligandai;
- X-ligandų labilumą lemia metalo kietumas/minkštumas ir pobūdis;
- „Numanomi“ ligandai yra labai labilūs: tirpikliai ir tilteliai dimeriuose ir klasteriuose, kad jų buvimas koordinavimo sferoje dažnai visiškai nepaisomas, o struktūros be jų nubrėžiamos su formaliai nesočiąja koordinavimo sfera;
- Dihapto ligandai, pavyzdžiui, alkenai ir alkinai, elgiasi kaip tipiški L ligandai: paprastai jie yra gana labilūs;
- didesnio haptiškumo ligandai retai būna labilūs, bet jei polihapto ligandas gali pakeisti prisijungimo prie monohapto būdą, jis tampa labilesnis, pvz., taip elgiasi η 3 -alilai;
- chelatiniai ligandai, sudarantys 5 ir 6 narių chelatinius žiedus, yra stabilūs, o chelatai su mažesniais arba didelis skaičius ciklo atomai yra labilūs, bent viename centre (atsidaro chelato žiedas ir ligandas lieka kaboti kaip paprastas). Pavyzdžiui, taip elgiasi acetatas;
Koordinaciniu požiūriu stabilūs ligandai
Pakartokime viską dar kartą, tik iš kitos pusės
Metalų koordinavimo sferoje paprastai išsaugomi (koordinaciškai stabilūs):
- 5 ir 6 narių chelatoriai;
- polihapto-ligandai: norint išmušti ciklopentadienilus ar benzeną (arenus) iš koordinavimo sferos, reikia naudoti visokius specialius būdus – jie tiesiog neišeina, dažnai atlaiko net ir ilgą kaitinimą;
- ligandai, prijungti prie metalo, turintys didelę π donoro efekto dalį (atgalinė donorystė);
- vėlyvųjų pereinamųjų metalų minkštieji ligandai;
- „paskutinis“ ligandas koordinavimo sferoje.
Paskutinė sąlyga atrodo keistai, bet įsivaizduokite kompleksą, kuriame yra daug skirtingų ligandų, tarp kurių nėra visiškai stabilių (nėra chelatorių ar polihapto-ligandų). Tada reakcijose ligandai pasikeis, santykinai tariant, santykinio labilumo tvarka. Mažiausiai labilus ir paskutinis išlikęs. Šis triukas pasitaiko, pavyzdžiui, kai naudojame paladžio fosfino kompleksus. Fosfinai yra santykinai stabilūs ligandai, tačiau kai jų daug, o metale gausu elektronų (d 8, d 10), jie vienas po kito užleidžia vietą aktoriams ligandams. Tačiau paskutinis fosfino ligandas dažniausiai lieka koordinavimo sferoje, ir tai yra labai gerai, atsižvelgiant į reakcijas, kuriose dalyvauja šie kompleksai. Prie šio svarbaus klausimo grįšime vėliau. Čia yra gana tipiškas pavyzdys, kai Hecko reakcijoje iš pradinės paladžio fosfino komplekso koordinavimo sferos lieka tik vienas, „paskutinis“ fosfinas. Šis pavyzdys mus labai priartina prie svarbiausios pereinamųjų metalų kompleksų reakcijose sąvokos – ligandų valdymo sampratos. Tai aptarsime vėliau.

Permetalavimas
Vienus ligandus pakeičiant kitais, svarbu nepersistengti dėl įeinančio ligando reaktyvumo. Kai susiduriame su organinių molekulių reakcijomis, mums svarbu į koordinavimo sferą tiekti tiksliai vieną kiekvieno reagento molekulę. Jei vietoj vienos molekulės patenka dvi, yra didelė šalutinių reakcijų, susijusių su dviem identiškais ligandais, tikimybė. Reaktyvumo praradimas taip pat galimas dėl koordinacinės sferos prisotinimo ir negalėjimo į ją įvesti kitų ligandų, reikalingų numatomam procesui. Ši problema ypač dažnai iškyla, kai į koordinavimo sferą patenka stiprūs anijoniniai nukleofilai, pavyzdžiui, karbanionai. Siekiant to išvengti, naudojami mažiau reaktyvūs dariniai, kuriuose vietoj didelį jungties joniškumą lemiančio šarminio metalo katijono naudojami mažiau elektropozityvūs metalai ir metaloidai (cinkas, alavas, boras, silicis ir kt.), formuojantys. kovalentiniai ryšiai su nukleofiline dalimi . Tokių darinių reakcijos su pereinamojo metalo dariniais gamina ligandų pakeitimo produktus, iš esmės taip, lyg nukleofilas būtų anijoninėje formoje, tačiau dėl sumažėjusio nukleofiliškumo su mažiau komplikacijų ir be šalutinių reakcijų.
Tokios ligandų pakeitimo reakcijos paprastai vadinamos transmetalacija, siekiant pabrėžti akivaizdų faktą, kad nukleofilas tarsi keičia metalus – labiau elektroteigiamus į mažiau elektropozityvius. Todėl šiame pavadinime yra nemalonios šizofrenijos elemento – atrodė, kad jau sutarėme, kad į visas reakcijas žiūrėsime pereinamojo metalo požiūriu, bet staiga vėl jį praradome ir pažiūrėjome į šią reakciją ir tik į šią reakciją. nukleofilo požiūriu. Teks apsišarvuoti kantrybe, tokia terminija susiformavo ir priimta. Tiesą sakant, šis žodis grįžta į ankstyvąją metalo organinių junginių chemiją ir į tai, kad ličio arba organinio magnio junginių poveikis įvairių metalų halogenidams ir metaloidams yra vienas iš pagrindinių visų metaloorganinių junginių, pirmiausia intranscinių, sintezės būdų. , ir reakcija, kurią dabar svarstome pereinamųjų metalų koordinacinių junginių chemijoje – tik apibendrinimas senas metodas metalo organinė chemija, iš kurios visa tai išaugo.
![]()
Kaip vyksta transmetalacija?
Permetalavimas yra panašus į įprastą pakeitimą ir nepanašus. Panašu, kad jei nepereinamąjį organometalinį reagentą laikysime tiesiog karbanjonu su priešjonu, tai anglies ir nepereinamojo metalo jungtis yra joninė. Tačiau atrodo, kad ši mintis tinka tik labiausiai elektropozityviems metalams – magniui. Bet jau dėl cinko ir alavo ši mintis labai toli nuo tiesos.
Todėl į reakciją patenka dvi σ jungtys ir keturi atomai jų galuose. Dėl to susidaro dvi naujos σ jungtys ir keturi atomai jungiasi vienas su kitu skirtinga tvarka. Labiausiai tikėtina, kad visa tai vyksta vienu metu keturių narių pereinamojoje būsenoje, o pati reakcija yra suderinta, kaip ir daugelis kitų pereinamųjų metalų reakcijų. Elektronų ir orbitalių gausa, tinkanti visiems skoniams ir visų tipų simetrijoms, leidžia pereinamiesiems metalams vienu metu palaikyti ryšius pereinamosiose būsenose su keliais atomais.
Remetalizacijos atveju gauname specialų atvejį labai bendras procesas, kuri tiesiog vadinama σ-ryšio metateze. Nepainiokite jų tik su tikrąja olefinų ir acetileno metateze, kurios yra visavertės katalizinės reakcijos su savo mechanizmais. Šiuo atveju kalbame apie transmetalacijos mechanizmą arba kitą procesą, kurio metu vyksta kažkas panašaus.

Įvadas į darbą
Darbo aktualumas. Porfirinų kompleksai su metalais, esantys aukštoje oksidacijos būsenoje, gali daug efektyviau koordinuoti bazes nei M 2+ kompleksai ir sudaryti mišrius koordinacinius junginius, kuriuose centrinio metalo atomo pirmoje koordinacinėje sferoje kartu su makrocikliniu ligandu yra ir neciklinių acidoligandų. o kartais koordinuotos molekulės. Ligandų suderinamumo tokiuose kompleksuose problemos yra labai svarbios, nes porfirinai atlieka savo biologines funkcijas mišrių kompleksų pavidalu. Be to, grįžtamosios bazinių molekulių, pasižyminčių vidutiniškai didelėmis pusiausvyros konstantomis, pridėjimo (pernešimo) reakcijos gali būti sėkmingai naudojamos organinių izomerų mišiniams atskirti. kiekybinė analizė, aplinkosaugos ir medicinos reikmėms. Todėl metaloporfirinų (MP) papildomo koordinavimo pusiausvyrų kiekybinių charakteristikų ir stechiometrijos bei paprastų ligandų pakeitimo juose tyrimai naudingi ne tik požiūriu. teorinių žinių metaloporfirinų, kaip kompleksinių junginių, savybes, bet ir sprendžiant praktinę mažų molekulių ar jonų receptorių ir pernešėjų paieškos problemą. Iki šiol sistemingų labai įkrautų metalų jonų kompleksų tyrimų praktiškai nėra.
Darbo tikslas. Tikras darbas skirta mišrių porfirino turinčių kompleksų, turinčių didelį krūvį metalų katijonų Zr IV, Hf IV, Mo V ir W V, reakcijų su bioaktyviomis N-bazėmis: imidazolu (Im), piridinu (Py), pirazinu (Pyz), benzimidazolu tyrimams. (BzIm), molekulinių kompleksų stabilumo charakteristikos ir optinės savybės, laipsniškų reakcijos mechanizmų pagrindimas.
Mokslinė naujovė. Taikant modifikuoto spektrofotometrinio titravimo, cheminės kinetikos, elektroninės ir vibracinės sugerties bei 1H BMR spektroskopijos metodus, pirmą kartą buvo gautos termodinaminės charakteristikos ir stechiometriniai N-bazių reakcijų su metaloporfirinais mechanizmai su mišria koordinavimo sfera (X) n -2 MTPP (X – acidoligandas Cl - , OH) buvo pagrįsti - , O 2- , TPP - tetrafenilporfirino dianionas). Nustatyta, kad daugeliu atvejų metaloporfirino-bazės supramolekulių susidarymo procesai vyksta laipsniškai ir apima keletą grįžtamų ir lėtų negrįžtamų elementarių bazinių molekulių koordinavimo ir rūgščių ligandų pakeitimo reakcijų. Kiekvienam laipsniškų reakcijų etapui buvo nustatytos stechiometrijos, pusiausvyros ar greičio konstantos, lėtų reakcijų eilės pagal bazę, o produktai spektriškai apibūdinti (UV, matomas spektras tarpiniams produktams ir UV, matomas ir IR galutiniams produktams). Pirmą kartą gautos koreliacinės lygtys, leidžiančios numatyti supramolekulinių kompleksų stabilumą su kitomis bazėmis. Lygtys darbe naudojamos aptarti detalų OH - pakeitimo Mo ir W kompleksuose bazine molekule mechanizmą. Aprašytos MR savybės, dėl kurių jis yra perspektyvus naudoti biologiškai aktyvių bazių aptikimui, atskyrimui ir kiekybinei analizei, pvz., vidutiniškai didelis supramolekulinių kompleksų stabilumas, aiškus ir greitas optinis atsakas, žemas jautrumo slenkstis ir antrasis. cirkuliacijos laikas.
Praktinė darbo reikšmė. Molekulinių kompleksų susidarymo reakcijų kiekybiniai rezultatai ir stechiometrinių mechanizmų pagrindimas turi didelę reikšmę makroheterociklinių ligandų koordinacinei chemijai. Disertaciniame darbe matyti, kad mišrūs porfirino turintys kompleksai pasižymi dideliu jautrumu ir selektyvumu bioaktyvioms organinėms bazėms, per kelias sekundes ar minutes suteikia optinį atsaką, tinkamą praktiškai aptikti reakcijas su bazėmis – LOJ, vaistų ir maisto produktų komponentais, kurį rekomenduojama naudoti kaip bazinių jutiklių komponentus ekologijoje, Maisto pramone, medicina ir žemės ūkis.
Darbo aprobavimas. Apie darbo rezultatus pranešta ir aptarta adresu:
IX Tarptautinė konferencija apie sprendimo ir kompleksavimo sprendiniuose problemas, Ples, 2004; XII simpoziumas apie tarpmolekulines sąveikas ir molekulių konformacijas, Pushchino, 2004; Rusijos seminaro apie porfirinų ir jų analogų chemiją XXV, XXVI ir XXIX mokslinės sesijos, Ivanovas, 2004 ir 2006 m.; VI NVS šalių jaunųjų mokslininkų mokykla-konferencija porfirinų ir giminingų junginių chemijos tema, Sankt Peterburgas, 2005 m.; VIII mokslinė mokykla – organinės chemijos konferencija, Kazanė, 2005 m.; Visos Rusijos mokslinė konferencija „Natūralūs makrocikliniai junginiai ir jų sintetiniai analogai“, Syktyvkaras, 2007 m.; XVI tarptautinė cheminės termodinamikos konferencija Rusijoje, Suzdalis, 2007 m.; XXIII tarptautinė Chugajevo koordinacinės chemijos konferencija, Odesa, 2007 m.; Tarptautinė porfirinų ir ftalocianinų konferencija ISPP-5, 2008 m.; 38-oji tarptautinė koordinacinės chemijos konferencija, Izraelis, 2008 m.
Pagrindinė vandeniniuose tirpaluose vykstanti pakeitimo reakcija – vandens molekulių mainai (22) – ištirta daugybei metalų jonų (34 pav.). Vandens molekulių mainai metalo jonų koordinavimo sferoje su didžiąja vandens molekulių, esančių kaip tirpiklis, dalis vyksta labai greitai daugumos metalų, todėl tokios reakcijos greitis gali būti tiriamas daugiausia relaksacijos metodu. Šis metodas apima sistemos pusiausvyros sutrikdymą, pavyzdžiui, staigiai pakilus temperatūrai. Naujomis sąlygomis (daugiau aukštos temperatūros) sistema nebebus pusiausvyros. Tada išmatuojamas pusiausvyros greitis. Jei galite pakeisti tirpalo temperatūrą viduje 10-8 sek, tada galėsite išmatuoti reakcijos, kuriai atlikti reikia daugiau nei laiko, greitį 10-8 sek.
Taip pat galima išmatuoti koordinuotų vandens molekulių pakeitimo greitį įvairiuose metalų jonais ligandais SO 2-4, S 2 O 3 2-, EDTA ir kt. (26). Šios reakcijos greitis
priklauso nuo hidratuoto metalo jono koncentracijos ir nepriklauso nuo įeinančio ligando koncentracijos, todėl šių sistemų greičiui apibūdinti galima naudoti pirmosios eilės lygtį (27). Daugeliu atvejų tam tikro metalo jono reakcijos greitis (27) nepriklauso nuo įeinančio ligando (L) pobūdžio, nesvarbu, ar tai būtų H 2 O molekulės, ar SO 4 2-, S 2 O 3 2-, ar EDTA jonai.
Šis stebėjimas, kartu su tuo, kad šio proceso greičio lygtis neapima įtekančio ligando koncentracijos, rodo, kad šios reakcijos vyksta mechanizmu, kuriame lėtas etapas apima ryšį tarp metalo jonų ir vandens. Tada gautas junginys greičiausiai greitai koordinuoja netoliese esančius ligandus.
Vabzdys. Šio skyriaus 4 punkte nurodyta, kad didesnio krūvio hidratuoti metalų jonai, tokie kaip Al 3+ ir Sc 3+, lėčiau keičia vandens molekules nei M 2+ ir M + jonai; Tai leidžia manyti, kad jungčių nutrūkimas vaidina svarbų vaidmenį etape, kuris lemia viso proceso greitį. Šių tyrimų metu gautos išvados nėra įtikinamos, tačiau leidžia manyti, kad S N 1 procesai yra svarbūs hidratuotų metalų jonų pakeitimo reakcijose.
Bene labiausiai ištirti kompleksiniai junginiai yra kobalto (III) aminai. Dėl jų stabilumo, lengvo paruošimo ir lėtos reakcijos jie ypač tinka kinetiniams tyrimams. Kadangi šių kompleksų tyrimai buvo atliekami tik vandeniniuose tirpaluose, pirmiausia turėtume atsižvelgti į šių kompleksų reakcijas su tirpiklio molekulėmis – vandeniu. Nustatyta, kad apskritai amoniako arba aminų molekulės, koordinuojamos Co(III) jono, taip lėtai pakeičiamos vandens molekulėmis, kad paprastai svarstoma pakeisti ligandus, išskyrus aminus.
Buvo ištirtas (28) tipo reakcijų greitis ir nustatyta, kad jis yra pirmos eilės, palyginti su kobalto kompleksu (X yra vienas iš daugelio galimų anijonų).
Kadangi vandeniniuose tirpaluose H 2 O koncentracija visada yra apytikslė 55,5 mln, tada neįmanoma nustatyti vandens molekulių koncentracijos pasikeitimo įtakos reakcijos greičiui. Greičio lygtys (29) ir (30), skirtos vandeninis tirpalas eksperimentiškai neatskiriami, nes k tiesiog lygus k" = k". Todėl iš reakcijos greičio lygties neįmanoma pasakyti, ar H2O dalyvaus greičio nustatymo proceso etape. Atsakymas į klausimą, ar ši reakcija vyksta S N 2 mechanizmu, pakeičiant X joną H 2 O molekule, ar S N 1 mechanizmu, kuris pirmiausia apima disociaciją, o vėliau H 2 O molekulės pridėjimą, turi būti gauti naudojant kitus eksperimentinius duomenis.
Šią problemą galima išspręsti dviejų tipų eksperimentais. Hidrolizės greitis (vieno Cl jono pakeitimas vandens molekulėje) transas- + yra maždaug 10 3 kartus didesnis nei hidrolizės greitis 2+. Padidėjęs komplekso krūvis sustiprina metalo ir ligandų ryšius ir, atitinkamai, slopina šių jungčių skilimą. Taip pat reikėtų atsižvelgti į gaunamų ligandų pritraukimą ir pakeitimo reakcijos palengvinimą. Kadangi didėjant komplekso krūviui buvo nustatytas greičio sumažėjimas, šiuo atveju labiau tikėtinas disociacinis procesas (S N 1).
Kitas įrodymo metodas yra pagrįstas daugelio panašių kompleksų hidrolizės tyrimu transas- +. Šiuose kompleksuose etilendiamino molekulė pakeičiama panašiais diaminais, kuriuose vandenilio atomai prie anglies atomo yra pakeisti CH 3 grupėmis. Kompleksai, kuriuose yra pakeistų diaminų, reaguoja greičiau nei etilendiamino kompleksas. Vandenilio atomus pakeitus CH 3 grupėmis, ligando tūris padidėja, todėl metalo atomą sunkiau atakuoti kitu ligandu. Šios sterinės kliūtys sulėtina reakciją per S N 2 mechanizmą. Tūrių ligandų buvimas šalia metalo atomo skatina disociacinį procesą, nes pašalinus vieną iš ligandų sumažėja jų kaupimasis prie metalo atomo. Pastebėtas kompleksų su didelių gabaritų ligandais hidrolizės greičio padidėjimas yra geras įrodymas, kad reakcija vyksta pagal S N 1 mechanizmą.
Taigi, atlikus daugybę Co (II) acidoamino kompleksų tyrimų, paaiškėjo, kad acido grupių pakeitimas vandens molekulėmis yra disociacinis procesas. Kobalto atomo-ligando ryšys išplečiamas iki tam tikros kritinės vertės, kol vandens molekulės pradeda patekti į kompleksą. Kompleksuose, kurių krūvis yra 2+ ir didesnis, kobalto-ligando ryšį nutraukti labai sunku, o vandens molekulių patekimas pradeda vaidinti svarbesnį vaidmenį.
Nustatyta, kad acido grupė (X -) kobalto (III) komplekse pakeičiama kita grupe nei H2O molekulė (31), pirmiausia ją pakeičiant molekule.
tirpiklis – vanduo, po to jį pakeičiant nauja grupė Y(32).
Taigi daugelyje reakcijų su kobalto(III) kompleksais reakcijos greitis (31) yra lygus hidrolizės greičiui (28). Tik hidroksilo jonas skiriasi nuo kitų reagentų reaktyvumu su Co (III) aminais. Jis labai greitai reaguoja su kobalto(III) aminų kompleksais (apie 106 kartus greičiau nei vanduo) pagal reakcijos tipą. bazinė hidrolizė (33).
Nustatyta, kad ši reakcija yra pirmosios eilės pakeičiančio ligando OH - (34) atžvilgiu. Bendra antroji reakcijos eiga ir neįprastai greita reakcijos eiga rodo, kad OH-jonas yra išskirtinai efektyvus nukleofilinis reagentas Co (III) kompleksams ir kad reakcija vyksta per S N 2 mechanizmą, susidarant tarpiniam junginiui.
Tačiau šią OH savybę galima paaiškinti ir kitu mechanizmu [lygtys (35), (36)]. Reakcijoje (35) 2+ kompleksas elgiasi kaip rūgštis (pagal Brønstedą), sudarydamas + kompleksą, kuris yra amido-(turintis)-junginys - bazė, atitinkanti rūgštį 2+.
Tada reakcija vyksta per S N 1 mechanizmą (36), kad susidarytų penkių koordinačių tarpinis produktas, kuris toliau reaguoja su tirpiklio molekulėmis ir susidaro galutinis reakcijos produktas (37). Šis reakcijos mechanizmas atitinka antros eilės reakcijos greitį ir atitinka S N 1 mechanizmą. Kadangi reakcijos greičio nustatymo stadijoje dalyvauja bazinis konjugatas su pradiniu kompleksu – rūgštimi, šis mechanizmas yra pavadintas S N. 1CB.
Labai sunku nustatyti, kuris iš šių mechanizmų geriausiai paaiškina eksperimentinius stebėjimus. Tačiau yra įtikinamų įrodymų, patvirtinančių S N 1CB hipotezę. Geriausi argumentai šio mechanizmo naudai yra šie: oktaedriniai Co(III) kompleksai paprastai reaguoja per S N 1 disociacinį mechanizmą, ir nėra įtikinamų argumentų, kodėl OH - jonas turėtų tarpininkauti S N 2 procesui. hidroksilo jonas yra silpnas nukleofilinis reagentas reakcijose su Pt(II), todėl neįprastas jo reaktyvumas su Co(III) atrodo nepagrįstas. Reakcijos su kobalto (III) junginiais nevandeninėje terpėje yra puikus įrodymas, kad susidaro penkių koordinačių tarpiniai produktai, kuriuos suteikia S N 1 SV mechanizmas.
Galutinis įrodymas yra tai, kad nesant N-H jungčių Co(III) komplekse, jis lėtai reaguoja su OH-jonais. Tai, žinoma, rodo, kad komplekso rūgščių-šarmų savybės yra svarbesnės nei OH nukleofilinės savybės reakcijos greičiui." Ši amino Co(III) kompleksų bazinės hidrolizės reakcija iliustruoja faktą, kad kinetikos duomenys dažnai gali būti interpretuojamas ne vienaip, o Norint atmesti vieną ar kitą galimą mechanizmą, reikia atlikti gana subtilų eksperimentą.
Šiuo metu ištirtos daugelio oktaedrinių junginių pakeitimo reakcijos. Jei atsižvelgsime į jų reakcijos mechanizmus, labiausiai paplitęs yra disociacinis procesas. Šis rezultatas nėra netikėtas, nes šeši ligandai aplink centrinį atomą palieka mažai vietos, kad kitos grupės galėtų prie jo prisijungti. Yra tik keli pavyzdžiai, kai buvo įrodytas septynių koordinačių tarpinis junginys arba buvo aptikta įsiterpusio ligando įtaka. Todėl S N 2 mechanizmas negali būti visiškai atmestas kaip galimas kelias pakeitimo reakcijos oktaedriniuose kompleksuose.
Sąlygiškai cheminės reakcijos kompleksai skirstomi į mainų, redokso, izomerizacijos ir koordinuotus ligandus.
Pirminė kompleksų disociacija į vidinę ir išorinę sferas lemia išorinės sferos jonų mainų reakcijų atsiradimą:
X m + mNaY = Y m + mNaX.
Kompleksų vidinės sferos komponentai taip pat gali dalyvauti medžiagų apykaitos procesuose, kuriuose dalyvauja ir ligandai, ir komplekso formuotojas. Norėdami apibūdinti ligandų ar centrinio metalo jono pakeitimo reakcijas, naudokite K. Ingoldo pasiūlytus organinių junginių reakcijoms pavadinimus ir terminus (42 pav.), nukleofilinius. S N ir elektrofilinis S E pakaitalai:
Z + Y = z + X S N
Z + M" = z + M S E .
Pagal pakeitimo reakcijos mechanizmą jie skirstomi (43 pav.) į asociatyvinius ( S N 1 ir S E 1 ) ir disociatyvus ( S N 2 ir S E 2 ), skiriasi pereinamojo laikotarpio būsena su padidėjusiu ir sumažėjusiu koordinavimo skaičiumi.
Reakcijos mechanizmo klasifikavimas kaip asociatyvus arba disociatyvus yra sunkiai eksperimentiškai pasiekiama užduotis identifikuoti tarpinę medžiagą su sumažintu arba padidintu koordinavimo skaičiumi. Šiuo atžvilgiu reakcijos mechanizmas dažnai vertinamas remiantis netiesioginiais duomenimis apie reagentų koncentracijos įtaką reakcijos greičiui, reakcijos produkto geometrinės struktūros pokyčius ir kt.
Norint apibūdinti kompleksų ligandų pakeitimo reakcijų greitį, Nobelio premijos laureatas 1983 G. Taube (44 pav.) pasiūlė vartoti terminus „labilis“ ir „inertiškas“, priklausomai nuo ligando pakeitimo reakcijos laiko, trumpiau arba ilgiau nei 1 minutę. Sąvokos labilus arba inertiškos yra ligandų pakeitimo reakcijų kinetikos charakteristikos ir neturėtų būti painiojamos su termodinaminėmis kompleksų stabilumo ar nestabilumo charakteristikomis.
Kompleksų labilumas arba inertiškumas priklauso nuo kompleksą sudarončio jono ir ligandų pobūdžio. Pagal ligandų lauko teoriją:
1. Aštuonkampiai kompleksai 3 d pereinamieji metalai su valentingumo pasiskirstymu ( n -1) d elektronų vienai sigmai*(pvz ) atsipalaiduojantys MO yra labilūs.
4- (t 2g 6 e g 1) + H2O= 3- + CN - .
Be to, kuo mažesnė komplekso kristalinio lauko stabilizavimo energija, tuo didesnis jo labilumas.
2. Aštuonkampiai kompleksai 3 d pereinamieji metalai su laisva sigma* atlaisvinimas pvz orbitalės ir vienodas valentingumo pasiskirstymas ( n -1) d elektronai t 2 g orbitose (t 2 g 3, t 2 g 6) yra inertiški.
[Co III (CN) 6] 3- (t 2 g 6 e g 0) + H 2 O =
[Cr III (CN) 6] 3- (t 2 g 3 e g 0) + H 2 O =
3. Plano kvadratas ir oktaedras 4 d ir 5 d pereinamieji metalai, neturintys elektronų vienoje sigmoje* atsipalaiduojantys MO yra inertiški.
2+ + H2O =
2+ + H2O =
Ligandų prigimties įtaka ligandų pakeitimo reakcijų greičiui nagrinėjama „ligandų tarpusavio įtakos“ modelio rėmuose. Ypatingas ligandų tarpusavio įtakos modelio atvejis yra tas, kurį 1926 m. suformulavo I.I. Černiajevo trans-įtakos samprata (45 pav.) – „Ligando labilumas komplekse priklauso nuo translokuoto ligando pobūdžio“ – ir siūlome daugybę ligandų trans-įtakų: CO, CN -, C 2 H 4 > PR 3, H - > CH 3 -, SC (NH 2) 2 > C 6 H 5 -, NO 2 -, I -, SCN - > Br -, Cl - > py , NH 3 , OH - , H 2 O .
Trans-įtakos samprata leido mums pateisinti nykščio taisykles:
1. Peyrone taisyklė- dėl amoniako ar aminų poveikio tetrachlorplatinatui ( II ) kalis visada gaunamas dichlorodiamino platinos cis konfigūracija:
2 - + 2NH3 = cis - + 2Cl - .
Kadangi reakcija vyksta dviem etapais ir chlorido ligandas turi didelę trans įtaką, antrasis chlorido ligandas pakeičiamas amoniaku susidarant cis-[ Pt (NH 3 ) 2 Cl 2 ]:
2- + NH3 = -
NH3 = cis -.
2. Jergenseno taisyklė - vandenilio chlorido rūgštimi veikiant platinos tetramino chloridui ( II ) arba panašių junginių gaunama dichlorodiammineplatinos trans konfigūracija:
[ Pt (NH 3 ) 4 ] 2+ + 2 HCl = trans-[ Pt (NH 3 ) 2 Cl 2 ] + 2 NH 4 Cl .
Pagal ligandų trans-įtakų seriją, pakeitus antrąją amoniako molekulę chlorido ligandu, susidaro trans-[ Pt (NH 3 ) 2 Cl 2].
3. Kurnakovo tiokarbamido reakcija - įvairūs tiokarbamido reakcijos su geometriniais trans-[ Pt (NH 3 ) 2 Cl 2 ] ir cis- [ Pt ( NH 3 ) 2 Cl 2 ]:
cis - + 4Tio = 2+ + 2Cl - + 2NH3.
Skirtingas charakteris reakcijos produktai yra susiję su dideliu tiokarbamido trans poveikiu. Pirmasis reakcijų etapas yra tiokarbamido chlorido ligandų pakeitimas trans- ir cis-[ Pt (NH3)2(Tio)2]2+:
trans-[Pt (NH 3) 2 Cl 2 ] + 2 Tio = trans-[ Pt (NH 3) 2 (Tio) 2 ] 2+
cis - + 2Tio = cis - 2+.
Cis-[Pt(NH3)2 (Tio ) 2 ] 2+ dvi amoniako molekulės, esančios trans-pozicijoje į tiokarbamidą, toliau pakeičiamos, todėl susidaro 2+ :
cis - 2+ + 2Tio = 2+ + 2NH3.
In trans-[Pt (NH3) 2 (Tio ) 2 ] 2+ dvi amoniako molekulės, turinčios mažą trans-įtaką, yra viena kitos atžvilgiu trans-pozicijoje, todėl jų nepakeičia tiokarbamidas.
Trans įtakos modelius atrado I.I. Černiajevas tirdamas ligandų pakeitimo reakcijas kvadratinių plokščių platinos kompleksuose ( II ). Vėliau buvo įrodyta, kad ligandų trans-įtaka pasireiškia ir kitų metalų kompleksuose ( Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III )) ir kita geometrinė struktūra. Tiesa, skirtingų metalų ligandų trans-įtakos serijos yra šiek tiek skirtingos.
Reikėtų pažymėti, kad trans įtaka yra kinetinis poveikis- kuo didesnė tam tikro ligando trans įtaka, tuo greičiau jis pakeičiamas kitu ligandu, kuris yra trans padėtyje jo atžvilgiu.
Kartu su kinetiniu trans įtakos efektu, per vidurį XX amžiaus A.A. Grinbergas ir Yu.N. Kukuškinas nustatė ligando trans-įtakos priklausomybę L nuo ligando, esančio cis padėtyje, iki L . Taigi, pakeitimo reakcijos greičio tyrimas Cl- amoniakas platinos kompleksuose ( II):
[PtCl 4 ] 2- + NH 3 = [ PtNH 3 Cl 3 ] - + Cl - K = 0,42. 10 4 l/mol. Su
[ PtNH 3 Cl 3 ] - + NH 3 = cis-[ Pt (NH 3 ) 2 Cl 2 ] + Cl - K = 1,14. 10 4 l/mol. Su
trans-[ Pt (NH 3 ) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2,90 . 10 4 l/mol. Su
parodė, kad vienos ar dviejų amoniako molekulių buvimas pakeisto chlorido ligando cis padėtyje nuosekliai padidina reakcijos greitį. Šis kinetinis efektas vadinamas cis įtaka. Šiuo metu abu kinetiniai ligandų pobūdžio įtakos ligandų pakeitimo reakcijų greičiui (trans- ir cis-poveikis) yra sujungti į bendrą koncepciją. ligandų tarpusavio įtaka.
Ligandų tarpusavio įtakos poveikio teorinis pagrindimas yra glaudžiai susijęs su minčių apie cheminius ryšius sudėtinguose junginiuose plėtojimu. 30-aisiais XX amžiaus A.A. Greenbergas ir B.V. Nekrasovas svarstė transįtaką poliarizacijos modelio rėmuose:
1. Trans efektas būdingas kompleksams, kurių centrinis metalo jonas yra labai poliarizuojamas.
2. Ligandų transaktyvumą lemia ligando ir metalo jono abipusės poliarizacijos energija. Tam tikram metalo jonui ligando trans įtaką lemia jo poliarizacija ir atstumas nuo centrinio jono.
Poliarizacijos modelis atitinka eksperimentinius duomenis apie kompleksus su paprastais anijoniniais ligandais, tokiais kaip halogenidų jonai.
1943 metais A.A. Greenbergas iškėlė hipotezę, kad ligandų transaktyvumas yra susijęs su jų redukuojančiomis savybėmis. Elektronų tankio poslinkis iš perkelto ligando į metalą sumažina efektyvų metalo jono krūvį, dėl kurio susilpnėja cheminis ryšys su perkeltu ligandu.
Idėjų apie trans poveikį vystymasis siejamas su dideliu ligandų transaktyvumu, pagrįstu nesočiomis organinėmis molekulėmis, tokiomis kaip etilenas. Pt(C2H4)Cl3 ] - . Pasak Chatt ir Orgel (46 pav.), taip yra dėl topi-tokių ligandų datyvinė sąveika su metalu ir asociatyvus pakeitimo reakcijų mechanizmas perkeltų ligandų atžvilgiu. Koordinavimas su puolančio ligando metalo jonu Z veda prie penkių koordinačių trigonalinio bipiramidinio tarpinio junginio susidarymo, po kurio greitai pašalinamas paliekantis ligandas X. Tokio tarpinio junginio susidarymą palengvinapi-datyvinio ligando ir metalo ligandų sąveika Y , kuris sumažina metalo elektronų tankį ir sumažina pereinamosios būsenos aktyvacijos energiją, vėliau greitai pakeičiant ligandą X.
Kartu su p akceptorius (C2H4, CN-, CO ...) ligandai, sudarantys datyvinio metalo ligandą cheminis ryšys, turi didelę trans įtaką irsdonorų ligandai: H - , CH 3 - , C 2 H 5 - ... Tokių ligandų trans-įtaką lemia ligando X donoro-akceptoriaus sąveika su metalu, dėl kurios sumažėja jo elektronų tankis ir susilpnėja metalo ryšys su išeinančiu ligandu. Y.
Taigi ligandų padėtis trans-aktyvumo serijoje yra nustatoma pagal bendrą sigma- donoras ir pi-ligandų savybės - sigma- donoras ir pi-ligando akceptorinės savybės sustiprina jo trans-įtaką, tuo tarpupi-donoriniai susilpnėja. Kuris iš šių ligando ir metalo sąveikos komponentų vyrauja transefekte, sprendžiama remiantis reakcijos pereinamosios būsenos elektroninės struktūros kvantiniais cheminiais skaičiavimais.
Vienas iš kritiniai etapai metalo komplekso katalizėje substrato Y sąveika su kompleksu vyksta trimis mechanizmais:
a) Ligando pakeitimas tirpikliu. Šis etapas paprastai vaizduojamas kaip komplekso disociacija
Proceso esmė daugeliu atvejų yra ligando pakeitimas tirpikliu S, kuris vėliau lengvai pakeičiamas substrato molekule Y
b) naujo ligando prijungimas prie laisvos koordinatės, susidarant asocijuotajam junginiui, po kurio seka pakeisto ligando disociacija
c) Sinchroninis pakeitimas (S N 2 tipas) be tarpinio susidarymo
Pt(II) kompleksų atveju reakcijos greitis labai dažnai apibūdinamas dviejų takų lygtimi
Kur k S Ir k Y yra procesų, vykstančių reakcijose (5) (su tirpikliu) ir (6) su ligandu Y, greičio konstantos. Pavyzdžiui,

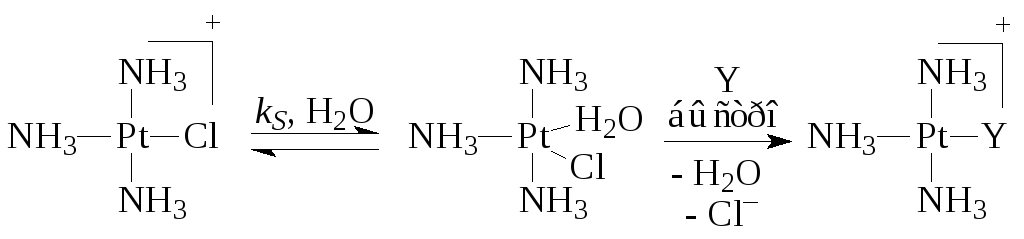
Paskutinis antrojo maršruto etapas yra trijų greitų elementarių etapų - Cl pašalinimo, Y pridėjimo ir H 2 O molekulės pašalinimo - suma.
Plokščiuose kvadratiniuose pereinamųjų metalų kompleksuose stebimas trans efektas, suformuluotas I. I. Chernyaev - LT įtaka ligando, esančio trans padėtyje LT ligandu, pakeitimo greičiui. Pt(II) kompleksams trans efektas padidėja ligandų serijoje:
H2O~NH3 Kinetinis trans-efektas ir termodinaminė trans-įtaka paaiškina galimybę susintetinti inertinius Pt(NH 3) 2 Cl 2 izomerinius kompleksus: Elektrofilinio vandenilio pakeitimo (S E) metalu reakcijos metalo koordinavimo sferoje ir jų atvirkštiniai procesai SH – H 2 O, ROH, RNH 2, RSH, ArH, RCCH. Tokio tipo reakcijose dalyvauja net H 2 ir CH 4 molekulės L įvedimo išilgai M-X jungties reakcijos X=R (organometalinio komplekso) atveju į M-R jungtį taip pat įvedamos metalo koordinuotos molekulės (L–CO, RNC, C 2 H 2, C 2 H 4, N 2, CO 2, O 2 ir kt. .). Įterpimo reakcija yra intramolekulinio nukleofilo atakos prieš arba koordinuotą molekulę rezultatas. Atvirkštinės reakcijos – - ir -eliminacijos reakcijos Oksidacinės pridėjimo ir redukcinės eliminacijos reakcijos M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4– Matyt, šiose reakcijose visada yra išankstinis pridėtos molekulės koordinavimas, tačiau to ne visada galima aptikti. Todėl laisvos vietos buvimas koordinavimo sferoje arba vieta, susijusi su tirpikliu, kuris lengvai pakeičiamas substratu, yra svarbus veiksnys, turintis įtakos metalų kompleksų reaktyvumui. Pavyzdžiui, Ni bis--alilo kompleksai yra geri kataliziškai aktyvių rūšių pirmtakai, nes dėl lengvo redukcinio bisalilo pašalinimo susidaro kompleksas su tirpikliu, vadinamasis. "plikas" nikelis. Tuščių sėdynių vaidmenį iliustruoja toks pavyzdys: Nukleofilinio ir elektrofilinio prisijungimo prie ir metalų kompleksų reakcijos Kaip katalizinių reakcijų tarpiniai produktai yra ir klasikiniai organiniai metaliniai junginiai, turintys M-C, M=C ir MC jungtis, ir neklasikiniai junginiai, kuriuose organinis ligandas koordinuojamas pagal 2 , 3 , 4 , 5 ir 6 -tipo, arba yra elektronų trūkumo struktūrų elementas – jungiantis CH 3 ir C 6 H 6 grupes, neklasikiniai karbidai (Rh 6 C(CO) 16, C(AuL) 5 +, C(AuL) 6 2+ ir kt.). Tarp specifinių klasikinių -organinių metalų junginių mechanizmų pažymime keletą mechanizmų. Taigi buvo nustatyti 5 metalo atomo elektrofilinio pakeitimo prie M-C jungties mechanizmai. elektrofilinis pakeitimas nukleofiline pagalba Papildymas-pašalinimas AdE(C) Pridėjimas prie C atomo sp 2 hibridizacijoje AdE(M) Oksidacinis priedas prie metalo Nukleofilinis pakeitimas anglies atome organinių metalų junginių demetalacijos reakcijose vyksta kaip redokso procesas: Galimas oksidatoriaus dalyvavimas šiame etape Toks oksidatorius gali būti CuCl 2, p-benzochinonas, NO 3 – ir kiti junginiai. Štai dar du pagrindiniai RMX etapai: M-C jungties hidrogenolizė ir M-C jungties homolizė Svarbi taisyklė, taikoma visoms sudėtingų ir organinių metalinių junginių reakcijoms ir susijusi su mažiausio judėjimo principu, yra Tolmano 16-18 elektronų apvalkalo taisyklė (2 skyrius).

Koordinuotų ligandų reakcijos








Metalo organinių junginių reakcijos








