Reaksjoner av koordinasjonsforbindelser forekommer alltid i koordinasjonssfæren til et metall med ligander bundet i det. Derfor er det åpenbart at for at noe i det hele tatt skal skje, må ligandene kunne falle inn i denne sfæren. Dette kan skje på to måter:
- et koordinativt umettet kompleks binder en ny ligand
- i en allerede fullført koordinasjonssfære erstattes en ligand med en annen.
Vi har allerede blitt kjent med den første metoden da vi diskuterte koordinasjonsumettethet og 18-elektronregelen. Vi tar for oss den andre her.
Ligander av enhver type kan substitueres i hvilken som helst kombinasjon
Men det pleier å fungere uuttalt regel– antall besatte koordineringsplasser endres ikke. Med andre ord endres ikke elektronantallet under substitusjon. Substitusjon av en type ligand med en annen er fullt mulig og forekommer ofte i virkeligheten. La oss bare ta hensyn til riktig håndtering av ladninger når vi endrer L-liganden til X-liganden og omvendt. Hvis vi glemmer dette, vil oksidasjonstilstanden til metallet endre seg, og erstatning av ligander er ikke en oksidasjonsreduksjonsprosess (hvis du finner eller kommer opp med et motsatt eksempel, gi meg beskjed - det vil automatisk bli kreditert rett bort, hvis jeg ikke kan bevise at du tok feil, og selv i dette tilfellet garanterer jeg et positivt bidrag til karma).
Substitusjon som involverer hapto-ligander
Med mer komplekse ligander er det ikke flere vanskeligheter - du trenger bare å huske en ganske åpenbar regel: antall ligandsteder (det vil si det totale antallet ligander eller X- eller L-ligandsentre) opprettholdes. Dette følger direkte av bevaring av elektrontelling. Her er selvinnlysende eksempler.

La oss ta hensyn til det siste eksemplet. Utgangsreagenset for denne reaksjonen er jerndiklorid FeCl2. Inntil nylig ville vi ha sagt: "Det er bare salt, hva har koordinasjonskjemi med det å gjøre?" Men vi vil ikke lenger tillate oss en slik uvitenhet. I kjemien til overgangsmetaller er det ingen "bare salter", alle derivater er koordinasjonsforbindelser, som alle betraktninger om elektrontelling, d-konfigurasjon, koordinasjonsmetning osv. gjelder. Jerndiklorid, slik vi er vant til å skrive det, skulle vise seg å være et Fe(2+)-kompleks av type MX 2 med konfigurasjon d 6 og antall elektroner 10. Ikke nok! Fint? Tross alt har vi allerede funnet ut at ligander kan være implisitte. For å lage reaksjonen trenger vi et løsemiddel, og for slike reaksjoner er det mest sannsynlig THF. Oppløsningen av et krystallinsk jernsalt i THF skjer nettopp fordi donorløsningsmidlet opptar ledige rom, og energien til denne prosessen kompenserer for ødeleggelsen krystallgitter. Vi ville ikke være i stand til å oppløse dette "saltet" i et løsningsmiddel som ikke gir metallløsningstjenestene på grunn av Lewis-grunnlaget. I dette tilfellet, og i en million lignende, er løsning ganske enkelt en koordinasjonsinteraksjon. La oss skrive, bare for nøyaktighetens skyld, resultatet av solvatisering i form av FeX 2 L 4-komplekset, der to klorioner forblir i koordinasjonssfæren i form av to X-ligander, selv om de mest sannsynlig også er fortrengt av molekyler av donorløsningsmidlet med dannelse av et ladet kompleks FeL 6 2+. I dette tilfellet er det ikke så viktig. Uansett kan vi trygt anta at vi har et 18-elektronkompleks på både venstre og høyre side.
Substitusjon, addisjon og dissosiasjon av ligander er nært og uløselig knyttet
Hvis vi husker organisk kjemi, så var det to substitusjonsmekanismer ved et mettet karbonatom - SN1 og SN2. I det første skjedde substitusjonen i to trinn: den gamle substituenten forlot først, og etterlot en ledig orbital på karbonatomet, som deretter ble okkupert av en ny substituent med et par elektroner. Den andre mekanismen antok at avreise og ankomst ble utført samtidig, i samråd, og prosessen var ett-trinns.
I kjemien til koordinasjonsforbindelser er det fullt mulig å forestille seg noe lignende. Men en tredje mulighet dukker opp, som det mettede karbonatomet ikke hadde - først fester vi en ny ligand, så løsner vi den gamle. Det blir umiddelbart klart at dette tredje alternativet neppe er mulig hvis komplekset allerede har 18 elektroner og er koordinasjonsmettet. Men det er fullt mulig hvis antallet elektroner er 16 eller mindre, det vil si at komplekset er umettet. La oss umiddelbart huske den åpenbare analogien fra organisk kjemi– nukleofil substitusjon ved et umettet karbonatom (i en aromatisk ring eller ved et karbonylkarbon) skjer også først som tilsetning av en ny nukleofil, og deretter eliminering av den gamle.
Så hvis vi har 18 elektroner, skjer substitusjonen som en abstraksjon-addisjon (fans av "smarte" ord bruker begrepet dissosiativ-assosiativ eller ganske enkelt dissosiativ mekanisme). En annen måte ville kreve å utvide koordinasjonssfæren til et antall på 20 elektroner. Dette er ikke helt umulig, og slike alternativer vurderes noen ganger til og med, men det er definitivt veldig ulønnsomt, og hver gang i tilfelle mistanke om en slik vei, kreves det svært betydelig bevis. I de fleste av disse historiene konkluderte forskerne til slutt med at de hadde oversett eller gått glipp av noe, og den assosiative mekanismen ble avvist. Så hvis det opprinnelige komplekset har 18 elektroner, må først en ligand forlate, deretter må en ny ta dens plass, for eksempel:

Hvis vi ønsker å introdusere en hapto-ligand som okkuperer flere steder i koordinasjonssfæren, må vi først forlate dem alle. Som regel skjer dette bare under ganske strenge forhold, for eksempel for å erstatte tre karbonyler i kromkarbonyl med η 6-benzen, oppvarmes blandingen under trykk i mange timer, og frigjør det frigjorte karbonmonoksidet fra tid til annen. Selv om diagrammet viser dissosiasjonen av tre ligander med dannelsen av et svært umettet kompleks med 12 elektroner, skjer reaksjonen mest sannsynlig i etapper, og etterlater en karbonyl om gangen, og benzen kommer inn i sfæren, og øker gradvis haptisiteten, gjennom trinn minus CO - digapto - minus en til CO - tetrahapto - minus en mer CO - heksagapto, slik at det ikke oppnås mindre enn 16 elektroner.

Så hvis vi har et kompleks med 16 elektroner eller mindre, så fortsetter erstatningen av liganden mest sannsynlig som en addisjonseliminering (for de som liker dyptklingende ord: assosiativ-dissosiativ eller rett og slett assosiativ): den nye liganden kommer først , så går den gamle. To åpenbare spørsmål dukker opp: hvorfor forlater den gamle liganden, fordi 18 elektroner er veldig gode, og hvorfor ikke gjøre det motsatte i dette tilfellet, som i 18-elektronkomplekser. Det første spørsmålet er enkelt å svare på: hvert metall har sine egne vaner, og noen metaller, spesielt sene, med nesten helt fylte d-skall, foretrekker 16-elektronantallet og de tilsvarende strukturtypene, og kaster derfor ut den ekstra liganden , og går tilbake til favorittkonfigurasjonen. Noen ganger forstyrrer også den romlige faktoren saken; de eksisterende ligandene er store og den ekstra føler seg som en busspassasjer i rushtiden. Det er lettere å gå av og gå en tur enn å lide slik. Du kan imidlertid skyve ut en annen passasjer, la ham gå en tur, så går vi. Det andre spørsmålet er også enkelt - i dette tilfellet vil den dissosiative mekanismen først måtte gi et 14-elektronkompleks, og dette er sjelden gunstig.
Her er et eksempel. For variasjon, la oss erstatte X-liganden med en L-ligand, og vi vil ikke bli forvirret om oksidasjonstilstander og ladninger. Nok en gang: ved substitusjon endres ikke oksidasjonstilstanden, og hvis X-liganden har forlatt, må tapet kompenseres med ladningen på metallet. Hvis vi glemmer dette, vil oksidasjonstallet synke med 1, men dette er feil.

Og en merkelig ting til. En metall-pyridinbinding ble dannet på grunn av det ensomme paret på nitrogen. I organisk kjemi vil vi i dette tilfellet definitivt vise et pluss på pyridin-nitrogenet (for eksempel ved protonering eller dannelse av et kvartært salt), men vi gjør aldri dette i koordineringskjemi med verken pyridin eller andre L-ligander. Dette er fryktelig irriterende for alle som er vant til det strenge og entydige systemet med å tegne strukturer i organisk kjemi, men du må venne deg til det, det er ikke så vanskelig.
Men det er ingen eksakt analog til SN2 i kjemien til koordinasjonsforbindelser; det er en fjern, men den er relativt sjelden og vi trenger den egentlig ikke.
Stabile og labile ligander
Vi kunne ikke snakket om mekanismene for ligandsubstitusjon i det hele tatt hvis ikke for en ekstremt viktig omstendighet som vi vil bruke mye: ligandsubstitusjon, det være seg assosiativ eller dissosiativ, forutsetter nødvendigvis dissosiasjonen av den gamle liganden. Og det er veldig viktig for oss å vite hvilke ligander som forlater lett og hvilke som forlater dårlig, og foretrekker å forbli i metallets koordinasjonssfære.
Som vi snart vil se, i enhver reaksjon forblir noen av liganden i koordinasjonssfæren og endres ikke. Slike ligander kalles vanligvis tilskuerligander (hvis du ikke vil ha slike enkle, "uvitenskapelige" ord, bruk engelsk ord tilskuer i lokal transkripsjon, tilskuer, ligand-tilskuer, men jeg ber deg, ikke tilskuer - dette er uutholdelig!). Og noen deltar direkte i reaksjonen, og blir til reaksjonsprodukter. Slike ligander kalles aktører (ikke aktører!), det vil si aktive. Det er helt klart at ligand-aktører enkelt må introduseres og fjernes i koordinasjonssfæren til metallet, ellers vil reaksjonen ganske enkelt sette seg fast. Men det er bedre å forlate tilskuerligander i koordinasjonssfæren av mange grunner, men i det minste for en så banal som behovet for å unngå unødvendig oppstyr rundt metallet. Det er bedre at bare ligander er aktører og nødvendige mengder kunne delta i riktig prosess. Hvis det er flere tilgjengelige koordinasjonssteder enn nødvendig, kan ekstra ligandaktører sitte på dem, og til og med de som vil delta i sidereaksjoner, noe som reduserer utbyttet av målproduktet og selektiviteten. I tillegg utfører tilskuerligander nesten alltid mange viktige funksjoner, for eksempel sikrer de løseligheten av komplekser, stabiliserer den riktige valenstilstanden til metallet, spesielt hvis det ikke er helt vanlig, hjelper individuelle stadier, gir stereoselektivitet, etc. Vi vil ikke tyde det ennå, fordi vi vil diskutere alt dette i detalj når vi kommer til spesifikke reaksjoner.
Det viser seg at noen av ligandene i koordinasjonssfæren skal være tett bundet og ikke utsatt for dissosiasjon og erstatning med andre ligander. Slike ligander kalles vanligvis koordinasjonsstabil . Eller rett og slett stabil, hvis det er klart fra konteksten at vi snakker om styrken til bindingene til liganden, og ikke om deres egen termodynamiske stabilitet, som ikke angår oss i det hele tatt.
Og ligander som lett og villig går inn og ut, og som alltid er klare til å vike for andre, kalles koordinasjonslabil , eller rett og slett labil, og her er det heldigvis ingen uklarheter.
Syklobutadien som ligand
Dette er nok det meste lysende eksempel det faktum at i koordinasjonssfæren kan et veldig ustabilt molekyl bli en utmerket ligand, og per definisjon koordinasjonsstabil, om ikke annet fordi hvis det våger å forlate den varme og koselige sfæren utenfor, venter det ikke noe godt (prisen på utgang vil være nøyaktig energien til antiaromatisk destabilisering).
Syklobutadien og dets derivater er de mest kjente eksemplene på antiaromatitet. Disse molekylene eksisterer bare når lave temperaturer, og i en svært forvrengt form - for å komme så langt som mulig fra antiaromatitet, blir syklusen forvrengt til et langstrakt rektangel, noe som fjerner delokalisering og maksimalt svekker konjugeringen av dobbeltbindinger (dette kalles ellers Jahn-Teller-effekten av 2. type: et degenerert system, og cyclobutadien square representerer er en degenerert biradikal, husk Frost-sirkelen - den er forvrengt og reduserer symmetri for å fjerne degenerasjonen).
Men i komplekser er cyklobutadien og substituerte cyklobutadiener utmerkede tetrahapto-ligander, og geometrien til slike ligander er nøyaktig en firkant, med identiske bindingslengder. Hvordan og hvorfor dette skjer er en egen historie, og ikke på langt nær så opplagt som det ofte blir gjort til.
Koordinasjonslabile ligander
Du må forstå at det ikke er noe armert betonggjerde med piggtråd og sikkerhetstårn mellom områdene med labile og stabile ligander. For det første kommer det an på metallet, og LMKO fungerer godt i denne sammenhengen. For eksempel foretrekker sene overgangsmetaller myke ligander, mens tidlige overgangsmetaller foretrekker harde. La oss si, jodid holder veldig tett til d 8-atomene til palladium eller platina, men kommer sjelden inn i koordinasjonssfæren til titan eller zirkonium i d 0-konfigurasjonen i det hele tatt. Men i mange metallkomplekser med mindre uttalte trekk manifesterer jodid seg som en fullstendig labil ligand, som lett gir etter for andre.
Annet likt:
- L-ligander er vanligvis mer labile enn X-ligander;
- labiliteten til X-ligander bestemmes av hardheten/mykheten og metallets natur;
- "implisitte" ligander er svært labile: løsemidler og broer i dimerer og klynger, så mye at deres tilstedeværelse i koordinasjonssfæren ofte blir fullstendig neglisjert og strukturer uten dem tegnes med en formelt umettet koordinasjonssfære;
- Dihapto-ligander, for eksempel alkener og alkyner, oppfører seg som typiske L-ligander: de er vanligvis ganske labile;
- ligander med større haptisitet er sjelden labile, men hvis en polyhapto-ligand kan endre bindingsmåten til mono-hapto, blir den mer labil, for eksempel oppfører η 3 -allyler på denne måten;
- chelatligander som danner 5- og 6-leddede chelatringer er stabile, og chelater med mindre eller et stort antall atomene i syklusen er labile, i det minste ved ett senter (chelatringen åpner seg og liganden forblir hengende som en enkel). Slik oppfører acetat seg for eksempel;
Koordinativt stabile ligander
La oss gjenta det hele igjen, bare på den andre siden
I koordinasjonssfæren til metaller er følgende generelt bevart (koordinasjonsstabil):
- 5- og 6-leddede chelatorer;
- polyhapto-ligander: for å slå cyklopentadienyler eller benzen (arener) ut av koordinasjonssfæren, må du bruke alle slags spesielle teknikker - de kommer bare ikke ut, tåler ofte til og med langvarig oppvarming;
- ligander bundet til et metall med høy andel π-donoreffekt (tilbakedonasjon);
- myke ligander for sene overgangsmetaller;
- "siste" ligand i koordinasjonssfæren.
Den siste tilstanden ser merkelig ut, men forestill deg et kompleks som har mange forskjellige ligander, blant dem er det ingen absolutt stabile (ingen chelatorer eller polyhapto-ligander). Deretter, i reaksjoner, vil liganden relativt sett endres i relativ labilitet. Den minst labile og den siste som er igjen. Dette trikset oppstår for eksempel når vi bruker palladiumfosfinkomplekser. Fosfiner er relativt stabile ligander, men når det er mange av dem, og metallet er rikt på elektroner (d 8, d 10), viker de etter hverandre for aktørligander. Men den siste fosfinliganden forblir vanligvis i koordinasjonssfæren, og dette er veldig bra med tanke på reaksjonene som disse kompleksene deltar i. Vi kommer tilbake til denne viktige saken senere. Her er et ganske typisk eksempel når bare ett, "siste" fosfin er igjen fra den innledende koordinasjonssfæren til palladiumfosfinkomplekset i Heck-reaksjonen. Dette eksemplet bringer oss veldig nær det viktigste konseptet i reaksjonene til overgangsmetallkomplekser - konseptet ligandkontroll. Vi diskuterer det senere.

Ommetallering
Når du erstatter noen ligander med andre, er det viktig å ikke overdrive reaktiviteten til den innkommende liganden. Når vi har å gjøre med reaksjoner av organiske molekyler, er det viktig for oss å levere nøyaktig ett molekyl av hver reaktant inn i koordinasjonssfæren. Hvis to molekyler kommer inn i stedet for ett, er det stor sannsynlighet for sidereaksjoner som involverer to identiske ligander. Et tap av reaktivitet er også mulig på grunn av metning av koordinasjonssfæren og umuligheten av å introdusere andre ligander som er nødvendige for den forventede prosessen. Dette problemet oppstår spesielt ofte når sterke anioniske nukleofiler, for eksempel karbanioner, introduseres i koordinasjonssfæren. For å unngå dette brukes mindre reaktive derivater, hvor det i stedet for alkalimetallkationet, som bestemmer den høye ionisiteten til bindingen, brukes mindre elektropositive metaller og metalloider (sink, tinn, bor, silisium, etc.), som danner kovalente bindinger med den nukleofile delen. Reaksjoner av slike derivater med overgangsmetallderivater produserer ligandsubstitusjonsprodukter, i prinsippet akkurat som om nukleofilen var i anionisk form, men på grunn av redusert nukleofilisitet med mindre komplikasjoner og ingen bireaksjoner.
Slike ligandsubstitusjonsreaksjoner kalles vanligvis transmetallering for å understreke det åpenbare faktum at nukleofilen ser ut til å endre metaller - mer elektropositive til mindre elektropositive. Dette navnet inneholder derfor et element av ubehagelig schizofreni - vi så ut til å allerede ha blitt enige om at vi skulle se på alle reaksjoner fra synspunktet til et overgangsmetall, men plutselig mistet vi det igjen og ser på denne reaksjonen og bare denne reaksjonen fra en nukleofils synspunkt. Du må smøre deg med tålmodighet, det er slik terminologien har utviklet seg og er akseptert. Faktisk går dette ordet tilbake til den tidlige kjemien til organometalliske forbindelser og til det faktum at virkningen av litium- eller organomagnesiumforbindelser på halogenider av forskjellige metaller og metalloider er en av hovedmetodene for syntese av alle organometalliske forbindelser, først og fremst intransisjonsforbindelser. , og reaksjonen som vi nå vurderer i kjemi av overgang- bare en generalisering gammel metode organometallisk kjemi, som det hele vokste fra.
![]()
Hvordan skjer transmetallering?
Remetallering er både lik konvensjonell substitusjon og ikke lik. Det ser ut som - hvis vi betrakter et organometallisk ikke-overgangsreagens som ganske enkelt et karbanion med et motion, så er karbon-ikke-overgangsmetallbindingen ionisk. Men denne ideen ser ut til å være sann bare for de mest elektropositive metallene - magnesium. Men allerede for sink og tinn er denne ideen veldig langt fra sannheten.
Derfor går to σ-bindinger og fire atomer i endene inn i reaksjonen. Som et resultat dannes to nye σ-bindinger og fire atomer binder seg til hverandre i en annen rekkefølge. Mest sannsynlig skjer alt dette samtidig i en overgangstilstand med fire medlemmer, og selve reaksjonen har en samordnet karakter, som så mange andre reaksjoner av overgangsmetaller. Overfloden av elektroner og orbitaler for bokstavelig talt enhver smak og alle typer symmetrier gjør overgangsmetaller i stand til samtidig å opprettholde bindinger i overgangstilstander med flere atomer.
Ved remetallisering får vi et spesielt tilfelle av svært generell prosess, som ganske enkelt kalles σ-bindingsmetatese. Ikke forveksle dem bare med sann metatese av olefiner og acetylener, som er fullverdige katalytiske reaksjoner med sine egne mekanismer. I dette tilfellet snakker vi om mekanismen for transmetallering eller en annen prosess der noe lignende oppstår.

Introduksjon til arbeidet
Arbeidets relevans. Komplekser av porfyriner med metaller i høye oksidasjonstilstander kan koordinere baser mye mer effektivt enn M 2+-komplekser og danne blandede koordinasjonsforbindelser der det i den første koordinasjonssfæren til det sentrale metallatomet, sammen med den makrosykliske liganden, er ikke-sykliske acidoligander. og noen ganger koordinerte molekyler. Spørsmål om ligandkompatibilitet i slike komplekser er ekstremt viktige, siden det er i form av blandede komplekser at porfyriner utfører sine biologiske funksjoner. I tillegg kan reversible addisjons(overførings)reaksjoner av basemolekyler, karakterisert ved moderat høye likevektskonstanter, med hell brukes til å separere blandinger av organiske isomerer, f. kvantitativ analyse, for miljømessige og medisinske formål. Derfor er studier av de kvantitative egenskapene og støkiometrien til likevektene for ytterligere koordinering på metalloporfyriner (MP) og substitusjon av enkle ligander i dem nyttige ikke bare fra synspunktet. teoretisk kunnskap egenskapene til metalloporfyriner som komplekse forbindelser, men også for å løse det praktiske problemet med å søke etter reseptorer og transportører av små molekyler eller ioner. Til dags dato er systematiske studier for komplekser av høyt ladede metallioner praktisk talt fraværende.
Målet med arbeidet. Virkelig arbeid er viet til studiet av reaksjonene av blandede porfyrinholdige komplekser av høyt ladede metallkationer Zr IV, Hf IV, Mo V og W V med bioaktive N-baser: imidazol (Im), pyridin (Py), pyrazin (Pyz), benzimidazol (BzIm), stabilitetsegenskaper og optiske egenskaper til molekylære komplekser, underbyggelse av trinnvise reaksjonsmekanismer.
Vitenskapelig nyhet. Ved å bruke metodene for modifisert spektrofotometrisk titrering, kjemisk kinetikk, elektronisk og vibrasjonsabsorpsjon og 1H NMR-spektroskopi, ble termodynamiske egenskaper oppnådd for første gang og de støkiometriske mekanismene for reaksjoner av N-baser med metalloporfyriner med en blandet koordinasjonssfære (X) n -2 MTPP (X - acidoligand Cl - , OH) ble dokumentert - , O 2- , TPP - tetrafenylporfyrin dianion). Det har blitt fastslått at i de aller fleste tilfeller fortsetter prosessene for dannelse av metalloporfyrin-base supramolekyler trinnvis og inkluderer flere reversible og langsomme irreversible elementære reaksjoner for koordinering av basemolekyler og substitusjon av sure ligander. For hvert trinn av trinnvise reaksjoner, støkiometri, likevekt eller hastighetskonstanter, ble rekkefølgen av langsomme reaksjoner basert på basen bestemt, og produktene ble spektralt karakterisert (UV, synlige spektre for mellomprodukter og UV, synlig og IR for sluttprodukter). For første gang er det oppnådd korrelasjonsligninger som gjør det mulig å forutsi stabiliteten til supramolekylære komplekser med andre baser. Ligningene brukes i arbeidet for å diskutere den detaljerte mekanismen for substitusjon av OH - i Mo- og W-kompleksene med et basemolekyl. Egenskapene til MR er beskrevet, noe som gjør det lovende for bruk i deteksjon, separasjon og kvantitativ analyse av biologisk aktive baser, slik som moderat høy stabilitet av supramolekylære komplekser, en klar og rask optisk respons, en lav sensitivitetsterskel og en andre sirkulasjonstid.
Arbeidets praktiske betydning. Kvantitative resultater og underbyggelse av de støkiometriske mekanismene for molekylære kompleksdannelsesreaksjoner er av betydelig betydning for koordineringskjemien til makroheterosykliske ligander. Avhandlingsarbeidet viser at blandede porfyrinholdige komplekser utviser høy sensitivitet og selektivitet overfor bioaktive organiske baser, i løpet av få sekunder eller minutter gir de en optisk respons egnet for praktisk påvisning av reaksjoner med baser - VOC, komponenter i legemidler og matprodukter, pga. som anbefales for bruk som komponenter av basesensorer i økologi, Mat industri, medisin og landbruk.
Godkjenning av arbeid. Resultatene av arbeidet ble rapportert og diskutert på:
IX Internasjonal konferanse om problemer med løsning og kompleksering i løsninger, Ples, 2004; XII Symposium om intermolekylære interaksjoner og konformasjoner av molekyler, Pushchino, 2004; XXV, XXVI og XXIX vitenskapelige sesjoner av det russiske seminaret om kjemi av porfyriner og deres analoger, Ivanovo, 2004 og 2006; VI Skolekonferanse for unge forskere fra CIS-landene om kjemien til porfyriner og relaterte forbindelser, St. Petersburg, 2005; VIII Scientific School - Conference on Organic Chemistry, Kazan, 2005; All-russisk vitenskapelig konferanse "Naturlige makrosykliske forbindelser og deres syntetiske analoger", Syktyvkar, 2007; XVI internasjonal konferanse om kjemisk termodynamikk i Russland, Suzdal, 2007; XXIII International Chugaev Conference on Coordination Chemistry, Odessa, 2007; Internasjonal konferanse om porfyriner og ftalocyaniner ISPP-5, 2008; 38th International Conference on Coordination Chemistry, Israel, 2008.
Hovedsubstitusjonsreaksjonen i vandige løsninger, utveksling av vannmolekyler (22), er studert for et stort antall metallioner (fig. 34). Utvekslingen av vannmolekyler i koordinasjonssfæren til et metallion med hoveddelen av vannmolekyler som er tilstede som et løsningsmiddel skjer veldig raskt for de fleste metaller, og derfor kan hastigheten på en slik reaksjon hovedsakelig studeres ved hjelp av relaksasjonsmetoden. Metoden går ut på å forstyrre systemets likevekt, for eksempel ved en kraftig temperaturøkning. Under nye forhold (mer høy temperatur) systemet vil ikke lenger være i likevekt. Likevektshastigheten måles deretter. Hvis du kan endre temperaturen på løsningen innenfor 10 -8 sek, så kan du måle hastigheten på en reaksjon som krever mer enn en periode å fullføre 10 -8 sek.
Det er også mulig å måle substitusjonshastigheten av koordinerte vannmolekyler i ulike metallioner med ligander SO 2-4, S 2 O 3 2-, EDTA, etc. (26). Hastigheten på denne reaksjonen
avhenger av konsentrasjonen av det hydratiserte metallionet og er ikke avhengig av konsentrasjonen av den innkommende liganden, noe som gjør det mulig å bruke førsteordensligningen (27) for å beskrive hastigheten til disse systemene. I mange tilfeller avhenger ikke reaksjonshastigheten (27) for et gitt metallion av naturen til den innkommende liganden (L), enten det er H 2 O-molekyler eller SO 4 2-, S 2 O 3 2-, eller EDTA-ioner.
Denne observasjonen, kombinert med det faktum at hastighetsligningen for denne prosessen ikke inkluderer konsentrasjonen av den innflytende liganden, antyder at disse reaksjonene foregår ved en mekanisme der det langsomme trinnet innebærer å bryte bindingen mellom metallionet og vann. Den resulterende forbindelsen koordinerer sannsynligvis raskt nærliggende ligander.
I Sect. 4 i dette kapittelet ble det uttalt at mer høyt ladede hydratiserte metallioner, som Al 3+ og Sc 3+, utveksler vannmolekyler langsommere enn M 2+ og M + ioner; Dette gir grunn til å anta at brudd på obligasjoner spiller en viktig rolle i det stadiet som bestemmer hastigheten på hele prosessen. Konklusjonene som er oppnådd i disse studiene er ikke avgjørende, men de gir grunn til å tro at S N 1-prosesser er viktige i substitusjonsreaksjoner av hydratiserte metallioner.
Sannsynligvis de mest studerte komplekse forbindelsene er kobolt(III)-aminer. Deres stabilitet, enkle tilberedning og langsomme reaksjoner gjør dem spesielt egnet for kinetiske studier. Siden studier av disse kompleksene utelukkende ble utført i vandige løsninger, bør vi først vurdere reaksjonene til disse kompleksene med løsemiddelmolekyler - vann. Det ble funnet at generelt blir ammoniakk- eller aminmolekyler koordinert av Co(III)-ionet så sakte erstattet av vannmolekyler at erstatning av andre ligander enn aminer vanligvis vurderes.
Reaksjonshastigheten av type (28) ble studert og funnet å være av første orden i forhold til koboltkomplekset (X er en av mange mulige anioner).
Siden i vandige løsninger er konsentrasjonen av H 2 O alltid omtrentlig 55,5 M, da er det umulig å bestemme effekten av å endre konsentrasjonen av vannmolekyler på reaksjonshastigheten. Hastighetsligninger (29) og (30) for vandig løsning kan ikke skjelnes eksperimentelt, siden k ganske enkelt er lik k" = k". Derfor er det umulig å si fra reaksjonshastighetsligningen om H2O vil delta i det hastighetsbestemmende trinnet i prosessen. Svaret på spørsmålet om denne reaksjonen fortsetter ved S N 2-mekanismen med erstatning av X-ionet med et H 2 O-molekyl eller ved S N 1-mekanismen, som først involverer dissosiasjon etterfulgt av tilsetning av et H 2 O-molekyl, må innhentes ved bruk av andre eksperimentelle data.
Dette problemet kan løses ved to typer eksperimenter. Hydrolysehastighet (erstatning av ett Cl - ion per vannmolekyl) transe- + er omtrent 10 3 ganger høyere enn hydrolysehastigheten 2+. En økning i ladningen av komplekset fører til styrking av metall-ligandbindinger og følgelig til inhibering av spaltningen av disse bindingene. Tiltrekningen av innkommende ligander og tilretteleggingen av substitusjonsreaksjonen bør også tas i betraktning. Siden en reduksjon i rate ble funnet ettersom ladningen av komplekset økte, virker i dette tilfellet en dissosiativ prosess (S N 1) mer sannsynlig.
En annen metode for bevis er basert på studiet av hydrolysen av en serie av lignende komplekser transe- + . I disse kompleksene er etylendiaminmolekylet erstattet med lignende diaminer, hvor hydrogenatomene ved karbonatomet er erstattet med CH 3-grupper. Komplekser som inneholder substituerte diaminer reagerer raskere enn etylendiaminkomplekset. Å erstatte hydrogenatomer med CH 3-grupper øker volumet av liganden, noe som gjør det vanskeligere for metallatomet å bli angrepet av en annen ligand. Disse steriske hindringene bremser reaksjonen via S N 2-mekanismen. Tilstedeværelsen av voluminøse ligander nær metallatomet fremmer den dissosiative prosessen, siden fjerning av en av liganden reduserer deres akkumulering ved metallatomet. Den observerte økningen i hydrolysehastigheten av komplekser med voluminøse ligander er godt bevis på at reaksjonen fortsetter i henhold til S N 1 mekanismen.
Så, som et resultat av mange studier av Co(II) acidoaminkomplekser, viste det seg at erstatning av acidogrupper med vannmolekyler er en dissosiativ prosess i naturen. Koboltatom-ligandbindingen utvides til en viss kritisk verdi før vannmolekyler begynner å komme inn i komplekset. I komplekser med en ladning på 2+ og høyere er det svært vanskelig å bryte kobolt-ligandbindingen, og inntreden av vannmolekyler begynner å spille en viktigere rolle.
Det ble funnet at erstatningen av acidogruppen (X -) i kobolt(III)-komplekset med en annen gruppe enn H2O-molekylet, (31) først går gjennom dens erstatning med et molekyl
løsemiddel - vann, etterfulgt av å erstatte det med ny gruppe Y(32).
I mange reaksjoner med kobolt(III)-komplekser er således reaksjonshastigheten (31) lik hydrolysehastigheten (28). Bare hydroksylionet skiller seg fra de andre reagensene i sin reaktivitet med Co(III)-aminer. Det reagerer veldig raskt med aminkomplekser av kobolt(III) (omtrent 10 6 ganger raskere enn vann) i henhold til reaksjonstypen basisk hydrolyse (33).
Denne reaksjonen ble funnet å være første orden med hensyn til den substituerende liganden OH - (34). Den generelle andre rekkefølgen av reaksjonen og den uvanlig raske fremdriften av reaksjonen antyder at OH-ionet er et eksepsjonelt effektivt nukleofilt reagens for Co(III)-komplekser og at reaksjonen fortsetter via S N 2-mekanismen via dannelsen av et mellomprodukt.
Imidlertid kan denne egenskapen til OH - også forklares med en annen mekanisme [ligning (35), (36)]. I reaksjon (35) oppfører 2+-komplekset seg som en syre (ifølge Brønsted), og gir +-komplekset, som er amido-(inneholdende)-forbindelse - base tilsvarende syre 2+.
Reaksjonen fortsetter deretter via S N 1-mekanismen (36) for å danne et fem-koordinat mellomprodukt, som videre reagerer med løsemiddelmolekyler for å produsere det endelige reaksjonsproduktet (37). Denne reaksjonsmekanismen er i samsvar med hastigheten til en annenordens reaksjon og tilsvarer mekanismen S N 1. Siden reaksjonen i det hastighetsbestemmende stadiet involverer et basekonjugat til det opprinnelige komplekset - syren, gis denne mekanismen betegnelsen S N 1 CB.
Det er svært vanskelig å bestemme hvilken av disse mekanismene som best forklarer eksperimentelle observasjoner. Imidlertid er det overbevisende bevis for å støtte S N 1CB-hypotesen. De beste argumentene for denne mekanismen er som følger: oktaedriske Co(III)-komplekser reagerer generelt via den SN 1-dissosiative mekanismen, og det er ikke noe overbevisende argument for hvorfor OH - ion skal mediere S N 2-prosessen. hydroksylionet er et svakt nukleofilt reagens i reaksjoner med Pt(II), og derfor virker dets uvanlige reaktivitet med Co(III) urimelig. Reaksjoner med kobolt(III)-forbindelser i ikke-vandige medier gir utmerket bevis for dannelsen av fem-koordinat-mellomprodukter levert av S N 1 SV-mekanismen.
Det endelige beviset er det faktum at i fravær av N - H-bindinger i Co(III)-komplekset, reagerer det sakte med OH - ioner. Dette antyder selvfølgelig at syre-base-egenskapene til komplekset er viktigere enn de nukleofile egenskapene til OH for reaksjonshastigheten." Denne reaksjonen av den grunnleggende hydrolysen av amin Co(III)-komplekser illustrerer det faktum at kinetiske data kan ofte tolkes på mer enn én måte, og For å utelukke en eller annen mulig mekanisme er det nødvendig å gjennomføre et ganske subtilt eksperiment.
For tiden er substitusjonsreaksjoner av et stort antall oktaedriske forbindelser studert. Hvis vi vurderer reaksjonsmekanismene deres, er den vanligste den dissosiative prosessen. Dette resultatet er ikke uventet siden seks ligander etterlater liten plass rundt det sentrale atomet slik at andre grupper kan feste seg til det. Det er bare noen få eksempler hvor forekomsten av et syv-koordinat-mellomprodukt er påvist eller påvirkning av en mellomliggende ligand er påvist. Derfor kan S N 2-mekanismen ikke fullstendig avvises som mulig vei substitusjonsreaksjoner i oktaedriske komplekser.
Betinget kjemiske reaksjoner komplekser er delt inn i utveksling, redoks, isomerisering og koordinerte ligander.
Den primære dissosiasjonen av komplekser i den indre og ytre sfæren bestemmer forekomsten av utvekslingsreaksjoner av ytre sfæreioner:
Xm + mNaY = Ym + mNaX.
Komponenter av den indre sfæren av komplekser kan også delta i metabolske prosesser som involverer både ligander og kompleksdannende middel. For å karakterisere substitusjonsreaksjoner av ligander eller det sentrale metallionet, bruk betegnelsene og terminologien foreslått av K. Ingold for reaksjoner av organiske forbindelser (fig. 42), nukleofile S N og elektrofil S E-erstatninger:
Z + Y = z +X S N
Z + M"= z + MSE.
I henhold til mekanismen for substitusjonsreaksjonen er de delt (fig. 43) i assosiative ( S N 1 og S E 1 ) og dissosiativ ( S N 2 og S E 2 ), forskjellig i overgangstilstanden med økt og redusert koordinasjonstall.
Å klassifisere en reaksjonsmekanisme som assosiativ eller dissosiativ er en vanskelig eksperimentelt oppnåelig oppgave med å identifisere et mellomprodukt med redusert eller økt koordinasjonstall. I denne forbindelse blir reaksjonsmekanismen ofte bedømt på grunnlag av indirekte data om effekten av konsentrasjonen av reagenser på reaksjonshastigheten, endringer i den geometriske strukturen til reaksjonsproduktet, etc.
For å karakterisere hastigheten på ligandsubstitusjonsreaksjoner av komplekser, Nobelprisvinner 1983 G. Taube (fig. 44) foreslo å bruke begrepene "labil" og "inert" avhengig av tiden for ligandsubstitusjonsreaksjonen, mindre enn eller mer enn 1 minutt. Begrepene labil eller inert er kjennetegn ved kinetikken til ligandsubstitusjonsreaksjoner og må ikke forveksles med termodynamiske egenskaper for stabiliteten eller ustabiliteten til komplekser.
Labiliteten eller inertheten til kompleksene avhenger av naturen til det kompleksdannende ionet og ligandene. I samsvar med ligandfeltteori:
1. Oktaedriske komplekser 3 d overgangsmetaller med fordeling av valens ( n -1) d elektroner per sigma*(f.eks ) løsne MO-er er labile.
4- (t 2g 6 e g 1) + H 2 O= 3- + CN-.
Jo lavere stabiliseringsenergien er av kompleksets krystallfelt, desto større er dets labilitet.
2. Oktaedriske komplekser 3 d overgangsmetaller med fri sigma* løsne f.eks orbitaler og en jevn fordeling av valens ( n -1) d elektroner i t 2 g orbitaler (t 2 g 3, t 2 g 6) er inerte.
[Co III (CN) 6] 3- (t 2 g 6 e g 0) + H 2 O =
[Cr III (CN) 6] 3- (t 2 g 3 e g 0) + H 2 O =
3. Plano-square og oktaedral 4 d og 5 d overgangsmetaller som ikke har elektroner per sigma* løsne MO-er er inerte.
2+ + H20 =
2+ + H20 =
Påvirkningen av liganders natur på hastigheten av ligandsubstitusjonsreaksjoner vurderes innenfor rammen av modellen "gjensidig påvirkning av ligander". Et spesielt tilfelle av modellen for gjensidig påvirkning av ligander er den som ble formulert i 1926 av I.I. Chernyaevs konsept om transpåvirkning (fig. 45) - "labiliteten til liganden i komplekset avhenger av naturen til den trans-lokaliserte liganden" - og foreslår en rekke trans-påvirkninger av liganden: CO, CN -, C 2 H 4 > PR 3, H - > CH 3 -, SC (NH 2) 2 > C 6 H 5 -, NO 2 -, I -, SCN - > Br -, Cl - > py , NH3, OH-, H20.
Konseptet med transpåvirkning tillot oss å rettferdiggjøre tommelfingerreglene:
1. Peyrones styre- på grunn av virkningen av ammoniakk eller aminer på tetraklorplatinat ( II ) Kalium oppnås alltid diklordiaminplatina cis-konfigurasjon:
2- + 2NH3 = cis- + 2Cl-.
Siden reaksjonen går i to trinn og kloridliganden har stor transpåvirkning, skjer erstatningen av den andre kloridliganden med ammoniakk med dannelse av cis-[ Pt (NH3)2Cl2]:
2- + NH 3 = -
NH3 = cis-.
2. Jergensens regel - på virkningen av saltsyre på platinatetraminklorid ( II ) eller lignende forbindelser oppnås diklordi-aminplatina trans-konfigurasjon:
[Pt (NH3)4]2+ + 2 HCl = trans-[Pt (NH3)2Cl2] + 2 NH4Cl.
I samsvar med serien av trans-påvirkninger av ligander, fører erstatning av det andre ammoniakkmolekylet med en kloridligand til dannelsen av trans-[ Pt (NH3)2Cl2].
3. Kurnakovs tiourea-reaksjon - forskjellige produkter av reaksjonen av tiourea med geometriske isomerer av trans-[ Pt (NH 3 ) 2 Cl 2 ] og cis- [ Pt (NH 3 ) 2 Cl 2 ]:
cis - + 4 Thio = 2+ + 2Cl- + 2NH3.
Annen karakter reaksjonsprodukter er assosiert med den høye transpåvirkningen av tiourea. Det første trinnet av reaksjonene er erstatning av tioureakloridligander med dannelse av trans- og cis-[ Pt (NH 3 ) 2 ( Thio ) 2 ] 2+ :
trans-[Pt (NH 3) 2 Cl 2 ] + 2 Thio = trans-[ Pt (NH 3) 2 (Thio) 2 ] 2+
cis - + 2Thio = cis - 2+.
In cis-[Pt (NH3)2 (Thio ) 2 ] 2+ to ammoniakkmolekyler i transposisjon til tiourea gjennomgår ytterligere substitusjon, noe som fører til dannelsen 2+ :
cis - 2+ + 2tio = 2+ + 2NH3.
I trans-[Pt (NH3)2 (Thio ) 2 ] 2+ to ammoniakkmolekyler med liten transpåvirkning er plassert i transposisjon til hverandre og erstattes derfor ikke av tiourea.
Mønstrene for transpåvirkning ble oppdaget av I.I. Chernyaev når man studerer ligandsubstitusjonsreaksjoner i kvadratiske platinakomplekser ( II ). Deretter ble det vist at trans-påvirkningen av ligander også manifesterer seg i komplekser av andre metaller ( Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III )) og annen geometrisk struktur. Riktignok er rekken av trans-påvirkning av ligander for forskjellige metaller noe forskjellig.
Det bør bemerkes at transpåvirkning er kinetisk effekt- jo større transpåvirkning en gitt ligand har, jo raskere erstattes den av en annen ligand som er i transposisjon i forhold til den.
Sammen med den kinetiske effekten av transpåvirkning, i midten XX århundre e.Kr. Grinberg og Yu.N. Kukushkin etablerte avhengigheten av trans-innflytelsen av liganden L fra liganden lokalisert i cis-posisjon til L . Dermed studiet av hastigheten på substitusjonsreaksjonen Cl- ammoniakk i platinakomplekser ( II):
[PtCl 4 ] 2- + NH 3 = [ PtNH 3 Cl 3 ] - + Cl - K = 0,42. 10 4 l/mol. Med
[PtNH 3 Cl 3 ] - + NH 3 = cis- [ Pt (NH 3 ) 2 Cl 2 ] + Cl - K = 1,14. 10 4 l/mol. Med
trans-[ Pt (NH 3 ) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2,90. 10 4 l/mol. Med
viste at tilstedeværelsen av ett eller to ammoniakkmolekyler i cis-posisjonen til den erstattede kloridliganden fører til en konsekvent økning i reaksjonshastigheten. Denne kinetiske effekten kalles cis innflytelse. For tiden er begge kinetiske effekter av påvirkningen av naturen til liganden på hastigheten av ligandsubstitusjonsreaksjoner (trans- og cis-effekt) kombinert i et generelt konsept gjensidig påvirkning av ligander.
Den teoretiske underbyggelsen av effekten av gjensidig påvirkning av ligander er nært knyttet til utviklingen av ideer om kjemiske bindinger i komplekse forbindelser. På 30-tallet XX århundre e.Kr. Greenberg og B.V. Nekrasov vurderte transpåvirkningen innenfor rammen av polarisasjonsmodellen:
1. Transeffekten er typisk for komplekser hvis sentrale metallion er svært polariserbart.
2. Transaktiviteten til ligander bestemmes av energien til gjensidig polarisering av liganden og metallionet. For et gitt metallion bestemmes transpåvirkningen av liganden av dens polariserbarhet og avstand fra sentralionet.
Polarisasjonsmodellen er i samsvar med eksperimentelle data for komplekser med enkle anioniske ligander, slik som halogenidioner.
I 1943 ble A.A. Greenberg antok at transaktiviteten til ligander er relatert til deres reduserende egenskaper. Skiftet i elektrontetthet fra den trans-lokaliserte liganden til metallet reduserer den effektive ladningen til metallionet, noe som fører til en svekkelse av den kjemiske bindingen med den trans-lokaliserte liganden.
Utviklingen av ideer om transpåvirkning er assosiert med den høye transaktiviteten til ligander basert på umettede organiske molekyler som etylen i [ Pt(C2H4)Cl3 ] - . I følge Chatt og Orgel (fig. 46) skyldes dettepi-den dative interaksjonen av slike ligander med metallet og den assosiative mekanismen for substitusjonsreaksjoner for trans-lokaliserte ligander. Koordinering til metallionet til den angripende liganden Z fører til dannelsen av et fem-koordinat trigonalt bipyramidalt mellomprodukt etterfulgt av rask eliminering av den utgående liganden X. Dannelsen av et slikt mellomprodukt lettes avpi-dativ ligand-metallligand interaksjon Y , som reduserer elektrontettheten til metallet og reduserer aktiveringsenergien til overgangstilstanden med påfølgende rask erstatning av ligand X.
Sammen med s akseptor (C2H4, CN-, CO ...) ligander som danner en dativ metallligand kjemisk forbindelse, har høy transinnflytelse ogsdonorligander: H - , CH 3 - , C 2 H 5 - ... Trans-påvirkningen av slike ligander bestemmes av donor-akseptor-interaksjonen av ligand X med metallet, noe som senker elektrontettheten og svekker bindingen til metallet med den utgående liganden Y.
Dermed blir posisjonen til liganden i serien av transaktivitet bestemt av den kombinerte virkningen av sigma- giver og pi-egenskaper til ligander - sigma- giver og pi-akseptoregenskapene til liganden øker dens trans-innflytelse, menspi-givere svekkes. Hvilken av disse komponentene i ligand-metall-interaksjonen som dominerer i trans-effekten, bedømmes på grunnlag av kvantekjemiske beregninger av den elektroniske strukturen til reaksjonens overgangstilstand.
En av kritiske stadier i metallkomplekskatalyse skjer interaksjonen mellom substratet Y og komplekset gjennom tre mekanismer:
a) Erstatning av liganden med et løsningsmiddel. Dette stadiet er vanligvis avbildet som dissosiasjonen av komplekset
Essensen av prosessen er i de fleste tilfeller erstatning av liganden med et løsningsmiddel S, som deretter lett erstattes av et substratmolekyl Y
b) Festing av en ny ligand ved en fri koordinat med dannelsen av en assosiert etterfulgt av dissosiasjon av den erstattede liganden
c) Synkron substitusjon (type S N 2) uten mellomdannelse
Når det gjelder Pt(II)-komplekser, er reaksjonshastigheten veldig ofte beskrevet av toveisligningen
Hvor k S Og k Y er hastighetskonstantene for prosesser som skjer i reaksjoner (5) (med et løsningsmiddel) og (6) med ligand Y. For eksempel,

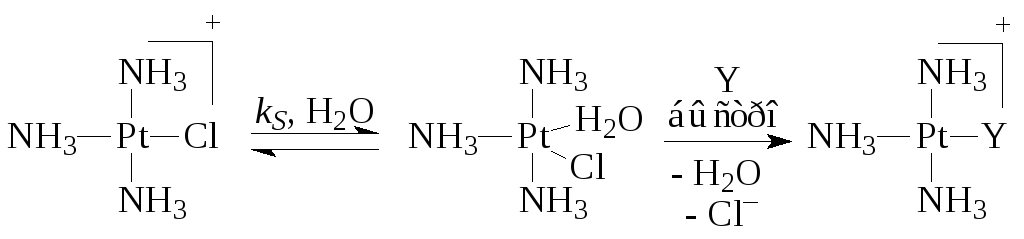
Den siste fasen av den andre ruten er summen av tre raske elementære stadier – eliminering av Cl –, tilsetning av Y og eliminering av H 2 O-molekylet.
I flate kvadratiske komplekser av overgangsmetaller observeres en transeffekt formulert av I.I. Chernyaev - påvirkningen av LT på substitusjonshastigheten til en ligand lokalisert i en transposisjon til LT-liganden. For Pt(II)-komplekser øker trans-effekten i rekken av ligander:
H 2 O~NH 3 Tilstedeværelsen av den kinetiske trans-effekten og termodynamisk trans-påvirkning forklarer muligheten for å syntetisere inerte isomere komplekser av Pt(NH 3) 2 Cl 2: Reaksjoner av elektrofil substitusjon (SE) av hydrogen med et metall i metallets koordinasjonssfære og deres inverse prosesser SH – H 2 O, ROH, RNH 2, RSH, ArH, RCCH. Selv H 2 og CH 4 molekyler deltar i reaksjoner av denne typen Reaksjoner ved introduksjon av L langs M-X-forbindelsen Ved X=R (organometallisk kompleks) introduseres også metallkoordinerte molekyler i M-R-bindingen (L–CO, RNC, C 2 H 2, C 2 H 4, N 2, CO 2, O 2, etc. .). Innsettingsreaksjonen er resultatet av et intramolekylært angrep av en nukleofil på et - eller -koordinert molekyl. Omvendte reaksjoner – - og -eliminasjonsreaksjoner Oksidative addisjons- og reduktive eliminasjonsreaksjoner M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4– Tilsynelatende er det i disse reaksjonene alltid foreløpig koordinering av det tilsatte molekylet, men dette kan ikke alltid oppdages. Derfor er tilstedeværelsen av et fritt sted i koordinasjonssfæren eller et sted assosiert med et løsningsmiddel som lett kan erstattes av et substrat en viktig faktor som påvirker reaktiviteten til metallkomplekser. For eksempel er bis--allylkomplekser av Ni gode forløpere for katalytisk aktive arter, siden på grunn av den enkle reduktive elimineringen av bis-allylen, oppstår et kompleks med løsningsmidlet, det såkalte. "bart" nikkel. Rollen til tomme seter illustreres av følgende eksempel: Reaksjoner av nukleofile og elektrofile tillegg til - og -komplekser av metaller Som mellomprodukter av katalytiske reaksjoner er det både klassiske organometalliske forbindelser med M-C, M=C og MC bindinger, og ikke-klassiske forbindelser der den organiske liganden er koordinert i henhold til 2 , 3 , 4 , 5 og 6 -type, eller er et element av elektronmangelfulle strukturer - brodannende CH 3 og C 6 H 6 grupper, ikke-klassiske karbider (Rh 6 C(CO) 16, C(AuL) 5 +, C(AuL) 6 2+ osv.). Blant de spesifikke mekanismene for klassiske -organometalliske forbindelser, merker vi flere mekanismer. Dermed er det etablert 5 mekanismer for elektrofil substitusjon av metallatomet ved M-C-bindingen. elektrofil substitusjon med nukleofil assistanse AdEaddition-eliminering AdE(C) Tilsetning til C-atomet i sp 2 hybridisering AdE(M) Oksidativ tilsetning til metall Nukleofil substitusjon ved karbonatomet i demetalliseringsreaksjoner av organometalliske forbindelser skjer som en redoksprosess: Mulig deltagelse av et oksidasjonsmiddel i dette stadiet Et slikt oksidasjonsmiddel kan være CuCl 2, p-benzokinon, NO 3 – og andre forbindelser. Her er ytterligere to elementære stadier som er karakteristiske for RMX: hydrogenolyse av M-C-bindingen og homolyse av M-C-bindingen En viktig regel som gjelder for alle reaksjoner av komplekse og organometalliske forbindelser og er assosiert med prinsippet om minste bevegelse er Tolmans 16-18 elektronskallregel (avsnitt 2).

Reaksjoner av koordinerte ligander








Reaksjoner av organometalliske forbindelser








