Reaktioner af koordinationsforbindelser forekommer altid i metallets koordinationssfære med liganderne bundet i det. Derfor er det indlysende, at for at der overhovedet kan ske noget, skal ligander kunne falde ind i denne sfære. Dette kan ske på to måder:
- det koordinativt umættede kompleks binder den nye ligand
- i en allerede afsluttet koordinationssfære skifter en ligand til en anden.
Vi har allerede stiftet bekendtskab med den første metode, da vi diskuterede koordinationsumættethed og 18-elektron reglen. Lad os gøre den anden her.
Ligander af enhver type kan substitueres i en hvilken som helst kombination
Men normalt er der en uudtalt regel – antallet af besatte koordinationspladser ændres ikke. Med andre ord ændrer substitution ikke elektronantallet. Substitutionen af en ligand af en type med en anden er ganske mulig og forekommer ofte i virkeligheden. Lad os kun være opmærksomme på den korrekte håndtering af ladninger, når L-liganden ændres til X-liganden og omvendt. Hvis vi glemmer dette, så vil graden af oxidation af metallet ændre sig, og udskiftningen af ligander er ikke en redoxproces (hvis du finder eller kommer med et grimt eksempel, så lad mig det vide - forskydningen vil være automatisk med det samme, hvis Jeg kan ikke bevise, at du tog fejl, hvorfor selv i dette tilfælde garanterer jeg et positivt bidrag til karma).
Substitution, der involverer hapto-ligander
Med mere komplekse ligander er der ikke flere vanskeligheder - du skal bare huske en ret indlysende regel: antallet af ligandsteder (det vil sige det samlede antal ligander eller ligandcentre af X- eller L-typer) er bevaret. Dette følger direkte af bevarelsen af elektronantallet. Her er nogle indlysende eksempler.

Lad os se på det sidste eksempel. Udgangsreagenset til denne reaktion er jerndichlorid FeCl2. Indtil for nylig ville vi have sagt: “Det er bare salt, hvad har koordinationskemien med det at gøre?”. Men vi vil ikke længere tillade os en sådan uvidenhed. I overgangsmetallernes kemi er der ingen "simpelthen salte", nogen derivater er koordinationsforbindelser, hvortil alle argumenter om elektrontælling, d-konfiguration, koordinationsmætning osv. er anvendelige. Jerndichlorid, som vi er vant til at skrive det, ville være et MX 2 type Fe(2+) kompleks med en d 6 konfiguration og 10 elektroner. Ikke nok! Bøde? Vi har jo allerede fundet ud af, at ligander er implicitte. For at lave en reaktion har vi brug for et opløsningsmiddel, og til sådanne reaktioner er det højst sandsynligt THF. Opløsningen af det krystallinske jernsalt i THF sker netop fordi donoropløsningsmidlet optager frie pladser, og energien i denne proces kompenserer for ødelæggelsen af krystalgitteret. Vi ville ikke være i stand til at opløse dette "salt" i et opløsningsmiddel, der ikke leverer metalopløsningstjenester på grund af Lewis-grundlæggende. I dette tilfælde, og i en million andre, der kan lide det, er løsning blot en koordinationsinteraktion. Lad os for en sikkerheds skyld skrive resultatet af solvatisering i form af et FeX 2 L 4 kompleks, hvor to chlorioner forbliver i koordinationssfæren i form af to X-ligander, selvom de højst sandsynligt også er fortrængt af donor opløsningsmiddel molekyler med dannelsen af et ladet kompleks FeL 6 2+. I dette tilfælde er det ikke så vigtigt. Og så, og så kan vi roligt antage, at vi har et 18-elektronkompleks til venstre og til højre.
Substitution, addition og dissociation af ligander er tæt og uløseligt forbundet
Hvis vi husker organisk kemi, så var der to substitutionsmekanismer ved et mættet carbonatom - SN1 og SN2. I det første skete substitutionen i to trin: den gamle substituent forlod først og efterlod en ledig orbital på carbonatomet, som blev efterfulgt af en ny substituent med et par elektroner. Den anden mekanisme antog, at afgang og ankomst udføres samtidigt, i samråd, og processen var en-trins.
I koordinationsforbindelsernes kemi er det meget muligt at forestille sig noget lignende. Men en tredje mulighed dukker op, som det mættede kulstofatom ikke havde - først vedhæfter vi en ny ligand, derefter kobler vi den gamle af. Det bliver straks klart, at denne tredje mulighed næppe er mulig, hvis komplekset allerede har 18 elektroner og er koordinativt mættet. Men det er meget muligt, hvis antallet af elektroner er 16 eller mindre, det vil sige, at komplekset er umættet. Lad os straks huske en åbenlys analogi fra organisk kemi - nukleofil substitution ved et umættet carbonatom (i en aromatisk ring eller carbonylcarbon) går også først som tilføjelse af en ny nukleofil, og derefter eliminering af den gamle.
Så hvis vi har 18 elektroner, så foregår substitutionen som en split-off-tilknytning (fans af "smarte" ord bruger udtrykket dissociativ-associativ eller blot dissociativ mekanisme). En anden måde ville kræve at udvide koordinationssfæren til et antal på 20 elektroner. Dette er ikke absolut umuligt, og sådanne muligheder overvejes nogle gange endda, men det er bestemt meget ufordelagtigt, og hver gang, hvis der er mistanke om en sådan vej, kræves der meget vægtige beviser. I de fleste af disse historier kom forskerne til sidst til den konklusion, at de overså eller ikke tog hensyn til noget, og den associative mekanisme blev afvist. Så hvis det oprindelige kompleks har 18 elektroner, så skal først en ligand forlade, så skal en ny komme i stedet for, for eksempel:

Hvis vi ønsker at introducere en hapto-ligand, der indtager flere positioner i koordinationssfæren, skal vi først frigive dem alle. Som regel sker dette kun under tilstrækkeligt strenge forhold, for eksempel for at erstatte tre carbonyler med η 6 -benzen i chromcarbonyl, opvarmes blandingen under tryk i mange timer, fra tid til anden udluftes det frigivne kulilte . Selvom skemaet viser dissociationen af tre ligander med dannelsen af et meget umættet kompleks med 12 elektroner, sker reaktionen i virkeligheden højst sandsynligt i etaper, efterlader en carbonyl, og benzen kommer ind i kuglen, hvilket gradvist øger hapticiteten gennem stadierne minus CO - dihapto - minus en mere CO - tetrahapto - minus en CO - hexagapto mere, så der ikke opnås mindre end 16 elektroner.

Så hvis vi har et kompleks med 16 elektroner eller derunder, så fortsætter ligandsubstitutionen højst sandsynligt som en additionsløsrivelse (for elskere af tankevækkende ord: associativ-dissociativ eller simpelthen associativ): den nye ligand kommer først, derefter den gamle blade. To åbenlyse spørgsmål opstår: hvorfor forlader den gamle ligand, fordi 18 elektroner er meget godt, og hvorfor ikke gøre det modsatte i dette tilfælde, som i 18-elektronkomplekser. Det første spørgsmål er let at besvare: hvert metal har sine egne vaner, og nogle metaller, især sene metaller med næsten helt fyldte d-skaller, foretrækker antallet af 16 elektroner og de tilsvarende strukturelle typer og kasserer derfor den ekstra ligand og vender tilbage til deres foretrukne konfiguration. Nogle gange forstyrrer den rumlige faktor også sagen, de allerede eksisterende ligander er store og den ekstra føler sig som en buspassager i myldretiden. Det er nemmere at stå af og gå rundt til fods end at lide sådan. Du kan dog skubbe en anden passager ud, lade ham gå en tur, så går vi. Det andet spørgsmål er også enkelt - i dette tilfælde skulle den dissociative mekanisme først give et 14-elektron kompleks, og det er sjældent gavnligt.
Her er et eksempel. Til en forandring vil vi erstatte X-liganden med en L-ligand, og vi vil ikke blive forvirrede i oxidationstilstande og ladninger. Endnu en gang: ved substituering ændres oxidationstilstanden ikke, og hvis X-liganden er væk, så skal tabet kompenseres af ladningen på metallet. Hvis vi glemmer dette, vil oxidationstilstanden falde med 1, hvilket ikke er sandt.

Og endnu en særhed. En metal-pyridin-binding blev dannet på grund af det enlige par på nitrogen. I organisk kemi vil vi i dette tilfælde nødvendigvis vise et plus på nitrogenet i pyridin (for eksempel under protonering eller dannelsen af et kvaternært salt), men vi gør det aldrig i koordineringskemi med hverken pyridin eller nogen anden L- ligander. Dette er frygtelig irriterende for alle, der er vant til det stramme og entydige system med at tegne strukturer i organisk kemi, men det skal man vænne sig til, det er ikke så svært.
Og der er ingen nøjagtig analog af SN2 i kemien af koordinationsforbindelser, der er en fjern, men den er relativt sjælden, og vi har ikke rigtig brug for den.
Stabile og labile ligander
Det ville være muligt slet ikke at tale om mekanismerne for ligandsubstitution, hvis ikke for en ekstremt vigtig omstændighed, som vi vil bruge meget: substitution af ligander, hvad enten det er associative eller dissociative, indebærer nødvendigvis dissociationen af den gamle ligand. Og det er meget vigtigt for os at vide, hvilke ligander der let forlader, og hvilke der forlader dårligt, og foretrækker at forblive i metallets koordinationssfære.
Som vi snart vil se, i enhver reaktion, forbliver nogle af liganderne i koordinationssfæren og ændrer sig ikke. Sådanne ligander kaldes normalt tilskuerligander (hvis du ikke vil have så enkle, "uvidenskabelige" ord, så brug det engelske ord spectator i den lokale transskription spectator, spectator ligand, men, jeg beder dig, ikke en tilskuer - det er uudholdeligt!) . Og en del deltager direkte i reaktionen og bliver til reaktionsprodukter. Sådanne ligander kaldes aktører (ikke aktører!), det vil sige skuespil. Det er helt klart, at ligand-aktørerne let skal indføres og fjernes i metallets koordinationssfære, ellers vil reaktionen simpelthen sætte sig fast. Men tilskuerligander er bedre tilbage i koordinationssfæren af mange grunde, men i det mindste af en sådan banal en som behovet for at undgå unødvendig ballade omkring metallet. Det er bedre, at kun ligander, aktører og i de nødvendige mængder kan deltage i den ønskede proces. Hvis der er flere tilgængelige koordinationssteder end nødvendigt, kan ekstra ligander-aktører sidde på dem, og endda dem, der vil deltage i sidereaktioner, hvilket reducerer udbyttet af målproduktet og selektiviteten. Derudover udfører tilskuerligander næsten altid mange vigtige funktioner, for eksempel at sikre opløseligheden af komplekser, stabilisere den korrekte valenstilstand af metallet, især hvis det ikke er helt normalt, hjælper individuelle trin, giver stereoselektivitet osv. Vi dechifrerer det ikke endnu, for vi vil diskutere alt dette i detaljer, når vi kommer til specifikke reaktioner.
Det viser sig, at nogle af liganderne i koordinationssfæren skal være stærkt bundet og ikke tilbøjelige til dissociation og substitution med andre ligander. Sådanne ligander kaldes koordinativt stabil . Eller ganske enkelt stabil, hvis det tydeligt fremgår af sammenhængen, at vi taler om ligandernes bindingsstyrke, og ikke om deres egen termodynamiske stabilitet, som bare ikke generer os overhovedet.
Og ligander, der let og villigt kommer ind og ud, og som altid er klar til at vige pladsen for andre, kaldes koordinativt labil , eller blot labil, og her er der heldigvis ingen uklarheder.
Cyclobutadien som en ligand
Her er nok det mest slående eksempel på, at et meget ustabilt molekyle i koordinationssfæren kan blive en fremragende ligand, og per definition koordinationsstabilt, om ikke andet fordi, hvis det begiver sig ud af den varme og hyggelige sfære, venter der intet godt. det (på bekostning af output er kun energien af anti-aromatisk destabilisering).
Cyclobutadien og dets derivater er de bedst kendte eksempler på antiaromaticitet. Disse molekyler eksisterer kun ved lave temperaturer og i en stærkt forvrænget form - for at komme så langt som muligt fra antiaromaticitet, forvrænges cyklussen til et aflangt rektangel, hvilket fjerner delokalisering og svækker konjugationen af dobbeltbindinger så meget som muligt (ellers , dette kaldes Jahn-Teller-effekten af 2. slags: degenereret system, og cyclobutadienfirkant er en degenereret diradikal, husk Frosts cirkel - forvrænget og reducerer symmetri for at fjerne degeneration).
Men i komplekser er cyclobutadien og substituerede cyclobutadiener fremragende tetrahapto-ligander, og geometrien af sådanne ligander er præcis en firkant med identiske bindingslængder. Hvordan og hvorfor det sker, er en særskilt historie, og langt fra så indlysende, som det ofte bliver præsenteret.
Koordinationslabile ligander
Du skal forstå, at der ikke er et armeret betonhegn med pigtråd og vagttårne mellem områderne med labile og stabile ligander. For det første afhænger det af metallet, og i denne sammenhæng fungerer GMKO godt. For eksempel foretrækker sene overgangsmetaller bløde ligander, mens tidlige overgangsmetaller foretrækker hårde ligander. For eksempel klæber iodid meget tæt til d 8 atomerne af palladium eller platin, men kommer sjældent endda ind i koordinationssfæren af titanium eller zirconium i d 0 konfigurationen. Men i mange metalkomplekser med ikke så udtalte træk manifesterer iodid sig som en fuldstændig labil ligand, der let giver plads til andre.
Alt andet lige:
- L-ligander er generelt mere labile end X-ligander;
- labiliteten af X-ligander bestemmes af hårdheden/blødheden og beskaffenheden af metallet;
- "implicitte" ligander er meget labile: opløsningsmidler og broer i dimerer og klynger, så meget, at deres tilstedeværelse i koordinationssfæren ofte negligeres helt, og strukturer tegnes uden dem med en formelt umættet koordinationssfære;
- digapto-ligander, såsom alkener og alkyner, opfører sig som typiske L-ligander: de er normalt ret labile;
- ligander med en højere lykke er sjældent labile, men hvis en poly-hapto ligand kan ændre bindingsmåden til en mono-hapto, bliver den mere labil, for eksempel opfører η 3 -allyler sig på denne måde;
- chelatligander, der danner 5- og 6-leddede chelatringe, er stabile, mens chelater med færre eller flere ringatomer er labile i det mindste ved ét center (chelatringen åbner sig, og liganden forbliver hængende som et simpelt). Sådan opfører for eksempel acetat sig;
Koordinationsstabile ligander
Lad os gøre det igen, men på den anden side
I koordinationssfæren af metaller bevares som regel følgende (er koordinatstabile):
- 5- og 6-leddede chelatorer;
- polyhapto-ligander: for at slå cyclopentadienyler eller benzen (arener) ud af koordinationssfæren, skal der bruges alle mulige specielle tricks - de kommer bare ikke ud på den måde, de tåler ofte selv langvarig opvarmning;
- ligander forbundet med metallet med en høj andel af π-donoreffekten (tilbagedonation);
- bløde ligander i sene overgangsmetaller;
- den "sidste" ligand i koordinationssfæren.
Den sidste tilstand ser mærkelig ud, men forestil dig et kompleks, der har mange forskellige ligander, blandt hvilke der ikke er nogen ubetinget stabile (ingen chelatorer og polygapto-ligander). Så vil liganderne i reaktionerne relativt set ændre sig i rækkefølgen af relativ labilitet. Den mindst labile og forbliver den sidste. Et sådant fokus opstår for eksempel, når vi bruger phosphinkomplekser af palladium. Fosfiner er relativt stabile ligander, men når der er mange af dem, og metallet er rigt på elektroner (d 8 , d 10), viger de, én efter én, for ligand-aktører. Men den sidste phosphinligand forbliver normalt i koordinationssfæren, og det er meget godt set ud fra de reaktioner, som disse komplekser deltager i. Vi vender tilbage til dette vigtige spørgsmål senere. Her er et ret typisk eksempel, hvor kun én "sidste" phosphin er tilbage fra den indledende koordinationssfære af palladiumphosphinkomplekset i Heck-reaktionen. Dette eksempel bringer os meget tæt på det vigtigste begreb i, begrebet ligandkontrol. Vi diskuterer senere.

Remetallisering
Når man erstatter en ligand med en anden, er det vigtigt ikke at overdrive det med reaktiviteten af den indkommende ligand. Når vi har at gøre med reaktioner af organiske molekyler, er det vigtigt for os at levere præcis et molekyle af hver af reagenserne til koordinationssfæren. Hvis to molekyler kommer ind i stedet for et, er der stor sandsynlighed for sidereaktioner, der involverer to identiske ligander. Et tab af reaktivitet er også muligt på grund af mætning af koordinationssfæren og umuligheden af at indføre andre ligander, der er nødvendige for den forventede proces. Dette problem opstår især ofte, når stærke anioniske nukleofiler, for eksempel carbanioner, indføres i koordinationssfæren. For at undgå dette anvendes mindre reaktive derivater, hvor der i stedet for en alkalimetalkation, som forårsager en høj bindingsionicitet, anvendes færre elektropositive metaller og metalloider (zink, tin, bor, silicium osv.), der danner kovalente bindinger med den nukleofile del. Reaktioner af sådanne derivater med overgangsmetalderivater giver i princippet ligandsubstitutionsprodukter nøjagtigt, som om nukleofilen var i den anioniske form, men på grund af reduceret nukleofilicitet med færre komplikationer og ingen sidereaktioner.
Sådanne ligandsubstitutionsreaktioner kaldes normalt transmetallering for at understrege det åbenlyse faktum, at nukleofilen ser ud til at ændre metaller - mere elektropositive til mindre elektropositive. Dette navn rummer således et element af ubehagelig skizofreni - vi har vist allerede været enige om, at vi vil se på alle reaktioner fra overgangsmetallets synspunkt, men pludselig brød vi sammen igen og ser på denne reaktion og kun denne reaktion fra en nukleofils synspunkt. Vi bliver nødt til at være tålmodige, sådan har terminologien udviklet sig, og sådan er den accepteret. Faktisk går dette ord tilbage til den tidlige kemi af organometalliske forbindelser og til det faktum, at virkningen af lithium- eller organomagnesiumforbindelser på halogenider af forskellige metaller og metalloider er en af de vigtigste metoder til syntese af enhver organometallisk, primært intransitiv, og den reaktion, som vi nu overvejer i kemien af koordinationsforbindelser af overgangsmetaller, er simpelthen en generalisering af den gamle metode for organometallisk kemi, hvorfra det hele voksede.
![]()
Hvordan foregår ommetallisering?
Ommetallering ligner både almindelig substitution og ikke. Det ser ud til, at hvis vi betragter et intransitivt organometallisk reagens som blot en carbanion med en modion, så er carbon-intransition metalbindingen ionisk. Men denne idé synes kun at være sand for de mest elektropositive metaller - for magnesium. Men allerede for zink og tin er denne idé meget langt fra sandheden.
Derfor indgår to σ-bindinger og fire atomer i deres ender i reaktionen. Som følge heraf dannes to nye σ-bindinger, og fire atomer er bundet til hinanden i en anden rækkefølge. Mest sandsynligt sker alt dette samtidigt i en fireleddet overgangstilstand, og selve reaktionen har en samordnet karakter, ligesom rigtig mange andre reaktioner af overgangsmetaller. Overfloden af elektroner og orbitaler for bogstaveligt talt enhver smag og alle slags symmetrier gør overgangsmetaller i stand til samtidig at opretholde bindinger i overgangstilstande med flere atomer.
Ved remetalisering får vi et særligt tilfælde af en meget generel proces, som blot kaldes σ-bindingsmetatese. Forveksle ikke kun med ægte metatese af olefiner og acetylener, som er fuldgyldige katalytiske reaktioner med deres egne mekanismer. I dette tilfælde taler vi om mekanismen for remetallisering eller en anden proces, hvor noget lignende opstår.

Introduktion til arbejdet
Arbejdets relevans. Komplekser af porphyriner med metaller i høje oxidationstilstande kan koordinere baser meget mere effektivt end M2+-komplekser og danne blandede koordinationsforbindelser, hvor der i den første koordinationssfære af det centrale metalatom sammen med den makrocykliske ligand er ikke-cykliske acidoligander, og nogle gange koordinerede molekyler. Spørgsmålene om kompatibilitet af ligander i sådanne komplekser er ekstremt vigtige, da det er i form af blandede komplekser, at porphyriner udfører deres biologiske funktioner. Derudover kan reaktioner med reversibel addition (overførsel) af basemolekyler, karakteriseret ved moderat høje ligevægtskonstanter, med succes anvendes til adskillelse af blandinger af organiske isomerer, til kvantitativ analyse, med henblik på økologi og medicin. Derfor er undersøgelser af de kvantitative egenskaber og støkiometri af yderligere koordinationsligevægte på metalloporphyriner (MP'er) og substitution af simple ligander i dem nyttige, ikke kun ud fra et synspunkt om teoretisk viden om egenskaberne af metalloporphyriner som komplekse forbindelser, men også til at løse det praktiske problem med at søge efter receptorer og bærere af små molekyler eller ioner. Indtil videre er der praktisk talt ingen systematiske undersøgelser af komplekser af højt ladede metalioner.
Målet med arbejdet. Dette arbejde er viet til undersøgelse af reaktionerne af blandede porphyrinholdige komplekser af højt ladede metalkationer Zr IV , Hf IV , Mo V og W V med bioaktive N-baser: imidazol (Im), pyridin (Py), pyrazin (Pyz) ), benzimidazol (BzIm), karakteriseringsstabilitet og optiske egenskaber af molekylære komplekser, underbyggelse af trinvise reaktionsmekanismer.
Videnskabelig nyhed. Metoder til modificeret spektrofotometrisk titrering, kemisk kinetik, elektronisk og vibrationsabsorption og 1H NMR-spektroskopi blev brugt for første gang til at opnå termodynamiske karakteristika og underbygge de støkiometriske mekanismer for reaktioner af N-baser med metalporphyriner med en blandet koordinationssfære (X) -, O 2-, TPP - tetraphenylporphyrin dianion). Det er blevet fastslået, at i langt de fleste tilfælde forløber processerne til dannelse af metalloporphyrin-base supramolekyler trinvis og omfatter adskillige reversible og langsomme irreversible elementære reaktioner af koordination af basemolekyler og substitution af acidoligander. For hvert trin af de trinvise reaktioner blev støkiometri, ligevægt eller hastighedskonstanter, basisrækkefølger af langsomme reaktioner bestemt, og produkterne blev spektralt karakteriseret (UV, synlige spektre for mellemprodukter og UV, synligt og IR for slutprodukter). For første gang er der opnået korrelationsligninger, som gør det muligt at forudsige stabiliteten af supramolekylære komplekser med andre baser. Ligningerne bruges i dette arbejde til at diskutere den detaljerede mekanisme for substitution af OH - i Mo- og W-komplekser med et basemolekyle. Egenskaberne ved MR er beskrevet, som bestemmer muligheden for at bruge dem til påvisning, adskillelse og kvantitativ analyse af biologisk aktive baser, såsom moderat høj stabilitet af supramolekylære komplekser, klar og hurtig optisk respons, lav følsomhedstærskel og en- anden cirkulationstid.
Arbejdets praktiske betydning. Kvantitative resultater og underbyggelse af de støkiometriske mekanismer af molekylære kompleksdannelsesreaktioner er afgørende for koordineringskemien af makroheterocykliske ligander. Afhandlingsarbejdet viser, at blandede porphyrinholdige komplekser udviser høj følsomhed og selektivitet med hensyn til bioaktive organiske baser, inden for få sekunder eller minutter giver de en optisk respons, der er egnet til praktisk påvisning af reaktioner med baser - VOC'er, komponenter af lægemidler og fødevarer , som derfor anbefales til brug som komponenter i basesensorer i økologi, fødevareindustri, medicin og landbrug.
Godkendelse af arbejde. Resultaterne af arbejdet blev rapporteret og diskuteret på:
IX International Conference on Problems of Solvation and Complex Formation in Solutions, Ples, 2004; XII Symposium om intermolekylære interaktioner og konformationer af molekyler, Pushchino, 2004; XXV, XXVI og XXIX videnskabelige sessioner af det russiske seminar om kemi af porphyriner og deres analoger, Ivanovo, 2004 og 2006; VI Skole-konference for unge forskere fra SNG-landene om kemien af porphyriner og relaterede forbindelser, St. Petersburg, 2005; VIII videnskabelig skole - konferencer om organisk kemi, Kazan, 2005; All-russisk videnskabelig konference "Naturlige makrocykliske forbindelser og deres syntetiske analoger", Syktyvkar, 2007; XVI international konference om kemisk termodynamik i Rusland, Suzdal, 2007; XXIII International Chugaev Conference on Coordination Chemistry, Odessa, 2007; International konference om porphyriner og phtalocyaniner ISPP-5, 2008; 38. internationale konference om koordinationskemi, Israel, 2008.
Hovedsubstitutionsreaktionen i vandige opløsninger - udvekslingen af vandmolekyler (22) - blev undersøgt for et stort antal metalioner (fig. 34). Udvekslingen af vandmolekyler i koordinationssfæren af en metalion med hovedparten af vandmolekyler, der er til stede som opløsningsmiddel, forløber meget hurtigt for de fleste metaller, og derfor blev hastigheden af en sådan reaktion hovedsageligt undersøgt ved afslapningsmetoden. Metoden består i at forstyrre systemets ligevægt, for eksempel ved en kraftig temperaturstigning. Under nye forhold (højere temperatur) vil systemet ikke længere være i ligevægt. Mål derefter ligevægtshastigheden. Hvis det er muligt at ændre temperaturen på opløsningen indenfor 10-8 sek, så er det muligt at måle hastigheden af en reaktion, der kræver et tidsinterval større end 10-8 sek.
Det er også muligt at måle substitutionshastigheden af koordinerede vandmolekyler i forskellige metalioner med ligander SO 2-4, S 2 O 3 2-, EDTA osv. (26). Hastigheden af en sådan reaktion
afhænger af koncentrationen af den hydratiserede metalion og afhænger ikke af koncentrationen af den indkommende ligand, hvilket gør det muligt at bruge førsteordensligningen (27) til at beskrive disse systemers hastighed. I mange tilfælde afhænger reaktionshastigheden (27) for en given metalion ikke af arten af den indkommende ligand (L), det være sig H 2 O-molekyler eller SO 4 2-, S 2 O 3 2- eller EDTA ioner.
Denne observation, og det faktum, at hastighedsligningen for denne proces ikke inkluderer koncentrationen af den indkommende ligand, antyder, at disse reaktioner forløber ved en mekanisme, hvor det langsomme trin er at bryde bindingen mellem metalionen og vand. Den resulterende forbindelse vil så sandsynligvis hurtigt koordinere nærliggende ligander.
I sek. 4 i dette kapitel blev det angivet, at mere højt ladede hydratiserede metalioner, såsom Al 3+ og Sc 3+, udveksler vandmolekyler langsommere end M 2+ og M+ ioner; dette giver grundlag for at antage, at bindingsbrud spiller en vigtig rolle i den fase, der bestemmer hastigheden af hele processen. Konklusionerne opnået i disse undersøgelser er ikke afgørende, men de giver grund til at tro, at S N 1 processer er vigtige i substitutionsreaktioner af hydratiserede metalioner.
Sandsynligvis de mest undersøgte komplekse forbindelser er cobalt(III)-aminer. Deres stabilitet, lette tilberedning og langsomme reaktioner med dem gør dem særligt velegnede til kinetiske undersøgelser. Da undersøgelserne af disse komplekser udelukkende blev udført i vandige opløsninger, er det først nødvendigt at overveje reaktionerne af disse komplekser med opløsningsmiddelmolekyler - vand. Det blev fundet, at ammoniak- eller aminmolekyler, der er koordineret af Co(III)-ionen, generelt erstattes så langsomt af vandmolekyler, at substitution af andre ligander end aminer normalt overvejes.
Reaktionshastigheden af type (28) blev undersøgt og fundet at være af første orden med hensyn til koboltkomplekset (X er en af mange mulige anioner).
Da koncentrationen af H 2 O i vandige opløsninger altid er ca 55,5 M, så er det umuligt at bestemme effekten af at ændre koncentrationen af vandmolekyler på reaktionshastigheden. Hastighedsligningerne (29) og (30) for en vandig opløsning kan eksperimentelt ikke skelnes, da k simpelthen er lig med k" = k". Derfor er det umuligt at sige ud fra reaktionshastighedsligningen, om H 2 O vil deltage i det trin, der bestemmer processens hastighed. Svaret på spørgsmålet, om denne reaktion forløber i henhold til S N 2-mekanismen med erstatning af X-ionen med H 2 O-molekylet eller ifølge S N 1-mekanismen, som først involverer dissociation efterfulgt af tilføjelse af H 2 O-molekylet , skal opnås ved hjælp af andre eksperimentelle data.
Dette problem kan løses ved to typer eksperimenter. Hydrolysehastighed (substitution af en Cl-ion pr. vandmolekyle) trance- + ca. 10 3 gange hydrolysehastigheden 2+ . En stigning i ladningen af komplekset fører til styrkelse af metal-ligand-bindingerne og følgelig til inhibering af brydningen af disse bindinger. Tiltrækningen af indkommende ligander og lettelsen af substitutionsreaktionen bør også tages i betragtning. Da et fald i hastigheden blev fundet, efterhånden som ladningen af komplekset steg, forekommer en dissociativ proces (S N 1) i dette tilfælde mere sandsynlig.
En anden måde at bevise på er baseret på undersøgelsen af hydrolysen af en række komplekser svarende til trance- + . I disse komplekser er ethylendiamin-molekylet erstattet af lignende diaminer, hvor hydrogenatomerne ved carbonatomet er erstattet af CH 3-grupper. Komplekser indeholdende substituerede diaminer reagerer hurtigere end ethylendiaminkomplekset. Udskiftning af hydrogenatomer med CH 3-grupper øger ligandens volumen, hvilket gør det vanskeligt for en anden ligand at angribe metalatomet. Disse steriske hindringer sænker reaktionen af S N 2-mekanismen. Tilstedeværelsen af voluminøse ligander nær metalatomet fremmer den dissociative proces, da fjernelse af en af liganderne reducerer deres akkumulering ved metalatomet. Den observerede stigning i hydrolysehastigheden af komplekser med voluminøse ligander er et godt bevis på, at reaktionen forløber ifølge S N 1 mekanismen.
Så som et resultat af talrige undersøgelser af Co(II) acidoaminkomplekser viste det sig, at udskiftningen af syregrupper med vandmolekyler er en dissociativ proces i naturen. Kobolt-atom-ligand-bindingen forlænges til en vis kritisk værdi, før vandmolekyler begynder at trænge ind i komplekset. I komplekser med en ladning på 2+ og højere er brydningen af kobolt-ligand-bindingen meget vanskelig, og indtrængen af vandmolekyler begynder at spille en vigtigere rolle.
Det blev fundet, at erstatningen af acidogruppen (X -) i cobalt(III)-komplekset med en anden gruppe end H 2 O-molekylet (31) først forløber gennem dens substitution med molekylet
opløsningsmiddel - vand, efterfulgt af dets udskiftning med en ny gruppe Y (32).
I mange reaktioner med cobalt(III)-komplekser er reaktionshastigheden (31) således lig med hydrolysehastigheden (28). Kun hydroxylionen adskiller sig fra andre reagenser med hensyn til reaktivitet med Co(III)-aminer. Det reagerer meget hurtigt med amminkomplekser af kobolt(III) (ca. 10 6 gange hurtigere end vand) alt efter reaktionstypen basisk hydrolyse (33).
Denne reaktion viste sig at være første orden med hensyn til den substituerende ligand OH- (34). Den overordnede anden orden af reaktionen og den usædvanligt hurtige progression af reaktionen tyder på, at OH-ionen er et usædvanligt effektivt nukleofilt reagens med hensyn til Co(III)-komplekser, og at reaktionen forløber via S N 2-mekanismen gennem dannelsen af et mellemprodukt .
Denne egenskab ved OH - kan dog også forklares med en anden mekanisme [ligning (35), (36)]. I reaktion (35) opfører kompleks 2+ sig som en syre (ifølge Brønsted), hvilket giver kompleks + , som er amido-(indeholdende)-forbindelse - en base svarende til en syre 2+.
Derefter forløber reaktionen ifølge S N 1 mekanismen (36) med dannelse af en fem-koordineret mellemforbindelse, som derefter reagerer med opløsningsmiddelmolekyler, hvilket fører til det endelige reaktionsprodukt (37). Denne reaktionsmekanisme er i overensstemmelse med andenordens reaktionshastighed og svarer til S N 1 mekanismen. Da reaktionen i det hastighedsbestemmende trin involverer en base konjugeret til det initiale syrekompleks, får denne mekanisme betegnelsen SN 1CB.
Det er meget vanskeligt at afgøre, hvilken af disse mekanismer der bedst forklarer eksperimentelle observationer. Der er dog stærke beviser, der understøtter S N 1CB-hypotesen. De bedste argumenter for denne mekanisme er som følger: oktaedriske Co(III)-komplekser reagerer generelt efter den dissociative S N 1-mekanisme, og der er ingen overbevisende argumenter for, hvorfor OH-ionen skulle forårsage S N 2-processen. hydroxylionen er et svagt nukleofilt reagens i reaktioner med Pt(II), og derfor virker dens usædvanlige reaktivitet med Co(III) urimelig. Reaktioner med cobalt(III)-forbindelser i ikke-vandige medier giver fremragende bevis for dannelsen af fem-koordinerede mellemprodukter tilvejebragt af S N 1 CB-mekanismen.
Det sidste bevis er det faktum, at i fravær af N - H-bindinger i Co(III)-komplekset, reagerer det langsomt med OH - ioner. Dette giver naturligvis grund til at tro, at kompleksets syre-base egenskaber er vigtigere end de nukleofile egenskaber af OH for reaktionshastigheden. Denne reaktion af den basiske hydrolyse af amminiske Co(III) komplekser er en illustration. af det faktum, at kinetiske data ofte kan fortolkes på mere end én måde, og For at udelukke denne eller hin mulige mekanisme er det nødvendigt at udføre et ret subtilt eksperiment.
På nuværende tidspunkt er substitutionsreaktioner af et stort antal oktaedriske forbindelser blevet undersøgt. Hvis vi betragter deres reaktionsmekanismer, så støder vi oftest på den dissociative proces. Dette resultat er ikke uventet, da de seks ligander efterlader lidt plads omkring det centrale atom, så andre grupper kan binde sig til det. Der kendes kun få eksempler, hvor forekomsten af et syv-koordinations-mellemprodukt er blevet bevist, eller effekten af en indtrængende ligand er blevet påvist. Derfor kan S N 2 mekanismen ikke fuldstændigt afvises som en mulig vej for substitutionsreaktioner i oktaedriske komplekser.
Konventionelt er de kemiske reaktioner af komplekser opdelt i udveksling, redox, isomerisering og koordinerede ligander.
Den primære dissociation af komplekser i de indre og ydre sfærer bestemmer forløbet af udvekslingsreaktionerne af ydre sfære-ioner:
Xm + mNaY = Ym + mNaX.
Komponenterne i kompleksernes indre sfære kan også deltage i udvekslingsprocesser, der involverer både liganderne og det kompleksdannende middel. For at karakterisere substitutionsreaktionerne af ligander eller den centrale metalion, notationen og terminologien foreslået af K. Ingold for reaktioner af organiske forbindelser (fig. 42), nukleofile S N og elektrofil S E udskiftninger:
Z + Y = z + XS N
Z + M"= z + MSE.
I henhold til mekanismen for substitutionsreaktionen er de opdelt (fig. 43) i associative ( S N 1 og S E 1 ) og dissociativ ( S N 2 og S E 2 ), som adskiller sig i overgangstilstanden med et øget og reduceret koordinationstal.
At tildele reaktionsmekanismen til associativ eller dissociativ er en vanskelig eksperimentelt opnåelig opgave med at identificere et mellemprodukt med et reduceret eller øget koordinationstal. I denne henseende bedømmes reaktionsmekanismen ofte på grundlag af indirekte data om virkningen af koncentrationen af reagenser på reaktionshastigheden, ændringer i reaktionsproduktets geometriske struktur osv.
For at karakterisere hastigheden af ligandsubstitutionsreaktioner i komplekser foreslog 1983-nobelpristageren G. Taube (fig. 44) at bruge udtrykkene "labil" og "inert" afhængigt af tidspunktet for ligandsubstitutionsreaktionen på mindre eller mere end 1 minut. Udtrykkene labil eller inert er karakteristika for kinetikken af ligandsubstitutionsreaktioner og bør ikke forveksles med termodynamiske karakteristika for kompleksers stabilitet eller ustabilitet.
Labiliteten eller inertheden af komplekserne afhænger af arten af den kompleksdannende ion og liganderne. Ifølge ligandfeltteorien:
1. Oktaedriske komplekser 3 d overgangsmetaller med en valensfordeling ( n -1) d elektroner pr. sigma*(f.eks ) af løsnede MO'er er labile.
4- (t 2g 6 e g 1) + H 2 O= 3- +CN-.
Desuden, jo lavere værdien af stabiliseringsenergien ved kompleksets krystalfelt er, jo større er dets labilitet.
2. Oktaedriske komplekser 3 d overgangsmetaller med fri sigma*hæve f.eks orbitaler og en ensartet fordeling af valens ( n -1) d elektroner i t 2 g orbitaler (t 2 g 3, t 2 g 6) er inerte.
[Co III (CN) 6] 3- (t 2 g 6 e g 0 ) + H 2 O =
[Cr III (CN) 6] 3- (t 2 g 3 e g 0 ) + H 2 O =
3. Plano-square og octahedral 4 d og 5d overgangsmetaller, der ikke har elektroner per sigma* løsne MO er inaktive.
2+ + H20 =
2+ + H20 =
Indflydelsen af liganders natur på hastigheden af ligandsubstitutionsreaktioner betragtes inden for rammerne af modellen "gensidig indflydelse af ligander". Et særligt tilfælde af modellen for gensidig påvirkning af ligander er formuleret i 1926 af I.I. Chernyaev begrebet trans-indflydelse (fig. 45) - "Labiliteten af liganden i komplekset afhænger af arten af den trans-lokaliserede ligand" - og foreslå en række trans-påvirkningsligander: CO , CN - , C 2 H 4 > PR 3 , H - > CH 3 - , SC (NH 2 ) 2 > C 6 H 5 - , NO 2 - , I - , SCN - > Br - , Cl - > py NH3, OH-, H20.
Begrebet trans-påvirkning gjorde det muligt at underbygge tommelfingerreglerne:
1. Peyronets regel- under påvirkning af ammoniak eller aminer på tetrachlorplatinat ( II ) Kalium opnås altid dichlordiaminplatinum cis-konfiguration:
2 - + 2NH3 \u003d cis - + 2Cl -.
Da reaktionen forløber i to trin, og chloridliganden har en stor transeffekt, sker substitutionen af den anden chloridligand med ammoniak med dannelse af cis-[ Pt (NH3)2Cl2]:
2- + NH 3 \u003d -
NH 3 \u003d cis -.
2. Jergensens styre - under påvirkning af saltsyre på platintetramminchlorid ( II ) eller lignende forbindelser opnås dichlordiaminplatin-transkonfiguration:
[Pt (NH3)4]2+ + 2 HCl = trans-[Pt (NH3)2Cl2] + 2 NH4Cl.
I overensstemmelse med rækken af trans-påvirkninger af ligander fører substitution af det andet ammoniakmolekyle til en chloridligand til dannelsen af trans-[ Pt (NH3)2Cl2].
3. Thiourea Kurnakov reaktion - forskellige produkter fra reaktionen af thiourinstof med geometriske isomerer af trans-[ Pt (NH 3 ) 2 Cl 2 ] og cis- [Pt (NH 3 ) 2 Cl 2 ]:
cis - + 4Thio \u003d 2+ + 2Cl - + 2NH 3.
Reaktionsprodukternes forskellige natur er forbundet med thiourinstofs høje transeffekt. Det første trin af reaktionerne er erstatningen af thiourinstofchloridligander med dannelsen af trans- og cis-[ Pt (NH3)2(Thio)2]2+:
trans-[ Pt (NH 3 ) 2 Cl 2 ] + 2 Thio = trans- [ Pt (NH 3 ) 2 (Thio ) 2 ] 2+
cis - + 2Thio = cis - 2+.
In cis-[Pt (NH3)2 (Thio ) 2 ] 2+ to ammoniakmolekyler trans til thiourinstof undergår yderligere substitution, hvilket fører til dannelsen 2+ :
cis - 2+ + 2Thio \u003d 2+ + 2NH 3.
I trans-[Pt (NH3)2 (Thio ) 2 ] 2+ to ammoniakmolekyler med en lille trans-effekt er placeret i trans-positionen til hinanden og erstattes derfor ikke af thiourinstof.
Mønstrene for trans-påvirkning blev opdaget af I.I. Chernyaev, når man studerer ligandsubstitutionsreaktioner i kvadratiske platinkomplekser ( II ). Efterfølgende blev det vist, at transeffekten af ligander også manifesterer sig i komplekser af andre metaller ( Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III )) og andre geometriske strukturer. Det er rigtigt, at rækken af trans-effekten af ligander for forskellige metaller er noget anderledes.
Det skal bemærkes, at trance indflydelse er kinetisk effekt- jo større trans-indflydelse denne ligand har, jo hurtigere er udskiftningen af en anden ligand, som er i forhold til den i trans-positionen.
Sammen med den kinetiske effekt af trans-påvirkning, i midten XX århundrede e.Kr. Grinberg og Yu.N. Kukushkin etablerede afhængigheden af transeffekten af liganden L fra liganden i cis-stilling til L . Således undersøgelsen af hastigheden af substitutionsreaktion Cl- ammoniak i platinkomplekser ( II):
[PtCl 4] 2- + NH 3 = [PtNH 3 Cl 3] - + Cl - K = 0,42. 104 l/mol. Med
[PtNH 3 Cl 3] - + NH 3 \u003d cis-[Pt (NH 3) 2 Cl 2] + Cl - K = 1,14. 104 l/mol. Med
trans-[Pt (NH 3 ) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2,90. 104 l/mol. Med
viste, at tilstedeværelsen af et eller to ammoniakmolekyler i cis-positionen til chloridliganden, der udskiftes, fører til en successiv stigning i reaktionshastigheden. Denne kinetiske effekt kaldes cis indflydelse. På nuværende tidspunkt er begge kinetiske virkninger af indflydelsen af liganders natur på hastigheden af ligandsubstitutionsreaktioner (trans- og cis-effekter) kombineret i et fælles koncept gensidig påvirkning af ligander.
Den teoretiske underbygning af virkningen af den gensidige påvirkning af ligander er tæt forbundet med udviklingen af ideer om den kemiske binding i komplekse forbindelser. I 30'erne XX århundrede e.Kr. Grinberg og B.V. Nekrasov betragtede trans-påvirkningen inden for rammerne af polarisationsmodellen:
1. Trans-effekten er karakteristisk for komplekser, hvis centrale metalion har en høj polariserbarhed.
2. Transaktiviteten af ligander bestemmes af den gensidige polarisationsenergi af liganden og metalionen. For en given metalion er trans-effekten af en ligand bestemt af dens polariserbarhed og afstand fra den centrale ion.
Polarisationsmodellen stemmer overens med eksperimentelle data for komplekser med simple anioniske ligander, for eksempel halogenidioner.
I 1943 A.A. Greenberg foreslog, at transaktiviteten af ligander er relateret til deres reducerende egenskaber. Skiftet af elektrontætheden fra den transaktive ligand til metallet reducerer den effektive ladning af metalionen, hvilket fører til en svækkelse af den kemiske binding med den trans-lokaliserede ligand.
Udviklingen af ideer om transeffekten er forbundet med den høje transaktivitet af ligander baseret på umættede organiske molekyler, såsom ethylen i [ Pt (C2H4)Cl3 ] - . Ifølge Chatt og Orgel (Fig. 46) skyldes dettepi-den dative interaktion af sådanne ligander med metallet og den associative mekanisme for substitutionsreaktioner for trans-lokaliserede ligander. Koordinering til metalionen af den angribende ligand Z fører til dannelsen af et fem-koordinat trigonalt-bipyramidalt mellemprodukt, efterfulgt af hurtig spaltning af den udgående ligand X. Dannelsen af et sådant mellemprodukt lettes ved atpi-dativ ligand-metalligand-interaktion Y , hvilket reducerer metallets elektrontæthed og reducerer aktiveringsenergien af overgangstilstanden med efterfølgende hurtig substitution af X-liganden.
Sammen med s acceptor (C2H4, CN-, CO ...) ligander der danner en dativ ligand-metal kemisk binding har en høj trans-påvirkning ogsdonorligander: H-, CH3-, C2H5- ... Trans-effekten af sådanne ligander bestemmes af donor-acceptor-interaktionen af liganden X med metallet, hvilket sænker dets elektrontæthed og svækker bindingen mellem metallet og den udgående ligand Y .
Ligandernes position i trans-aktivitetsrækken bestemmes således af den kombinerede virkning af sigma donor og pi-egenskaber af ligander - sigma- donor og pi-ligandens acceptoregenskaber øger dens trans-effekt, menspi-donor - svække. Hvilke af disse komponenter af ligand-metal-interaktionen, der er fremherskende i trans-effekten, bedømmes ud fra kvantekemiske beregninger af den elektroniske struktur af reaktionens overgangstilstand.
Et af de vigtigste trin i metalkomplekskatalyse, interaktionen af Y-substratet med komplekset, forløber via tre mekanismer:
a) Udskiftning af liganden med et opløsningsmiddel. Normalt er et sådant stadium afbildet som dissociationen af komplekset
Essensen af processen er i de fleste tilfælde udskiftningen af liganden L med opløsningsmidlet S, som derefter let erstattes af substratmolekylet Y
b) Vedhæftning af en ny ligand langs en fri koordinat med dannelsen af en associeret, efterfulgt af dissociation af den substituerede ligand
c) Synkron substitution (type S N 2) uden dannelse af et mellemprodukt
I tilfælde af Pt(II)-komplekser beskrives reaktionshastigheden meget ofte ved tovejsligningen
Hvor k S Og k Y er hastighedskonstanterne for de processer, der finder sted i reaktioner (5) (med opløsningsmiddel) og (6) med ligand Y. For eksempel,

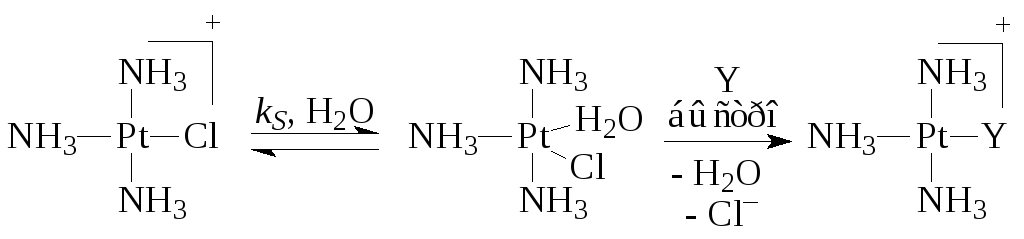
Det sidste trin af den anden rute er summen af tre hurtige elementære stadier - spaltning af Cl -, tilsætning af Y og eliminering af H 2 O-molekylet.
I plane kvadratiske komplekser af overgangsmetaller observeres en trans-effekt, formuleret af I.I. Chernyaev - virkningen af LT på substitutionshastigheden af en ligand, der er i en trans-position til LT-liganden. For Pt(II)-komplekser øges trans-effekten i rækken af ligander:
H2O~NH3 Tilstedeværelsen af den kinetiske trans-effekt og den termodynamiske trans-effekt forklarer muligheden for at syntetisere inerte isomere komplekser Pt(NH 3) 2 Cl 2: Reaktioner af elektrofil substitution (SE) af hydrogen med et metal i metallets koordinationssfære og deres omvendte processer SH - H 2 O, ROH, RNH 2, RSH, ArH, RCCH. Selv H 2 og CH 4 molekyler er involveret i reaktioner af denne type Indsættelsesreaktioner L ved binding M-X I tilfælde af X=R (et organometallisk kompleks) indføres også metalkoordinerede molekyler ved M-R-bindingen (L–CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 , etc.). Insertionsreaktioner er resultatet af et intramolekylært angreb af nukleofil X på et - eller -koordineret molekyle. Omvendte reaktioner - - og -elimineringsreaktioner Oxidative additions- og reduktive eliminationsreaktioner M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4– Tilsyneladende er der i disse reaktioner altid en foreløbig koordination af det vedhæftede molekyle, men dette er ikke altid muligt at fikse. Derfor er tilstedeværelsen af et frit sted i koordinationssfæren eller et sted forbundet med opløsningsmidlet, som let erstattes af substratet, en vigtig faktor, der påvirker reaktiviteten af metalkomplekser. For eksempel er bis--allylkomplekser af Ni gode forløbere for katalytisk aktive arter, da på grund af den lette reduktive eliminering af bis-allyl, et kompleks med et opløsningsmiddel, den såkaldte. bart nikkel. Rollen af ledige pladser illustreres af følgende eksempel: Reaktioner af nukleofil og elektrofil addition til - og -metalkomplekser Som mellemprodukter af katalytiske reaktioner findes der både klassiske organometalliske forbindelser med M-C, M=C og MC bindinger, såvel som ikke-klassiske forbindelser, hvor den organiske ligand er koordineret efter 2 , 3 , 4 , 5 og 6-typen, eller er et element af elektronmangelfulde strukturer - brodannende CH 3 og C 6 H 6 grupper, ikke-klassiske karbider (Rh 6 C (CO) 16, C (AuL) 5 +, C (AuL) ) 6 2+ osv.). Blandt de specifikke mekanismer for klassiske -organometalliske forbindelser bemærker vi flere mekanismer. Således er 5 mekanismer for elektrofil substitution af et metalatom ved M-C-bindingen blevet etableret. elektrofil substitution med nukleofil assistance AdEaddition-eliminering AdE(C)-binding til C-atom i sp2-hybridisering AdE(M) Oxidativ tilsætning til metal Nukleofil substitution ved carbonatomet i reaktionerne ved demetallisering af organometalliske forbindelser forekommer som en redoxproces: Det er muligt, at et oxidationsmiddel kan være involveret i dette trin. CuCl2, p-benzoquinon, NO3- og andre forbindelser kan tjene som et sådant oxidationsmiddel. Her er to mere elementære stadier, der er karakteristiske for RMX: M-C-bindingshydrogenolyse og homolyse af M-C-bindingen En vigtig regel vedrørende alle reaktioner af komplekse og organometalliske forbindelser og relateret til princippet om mindste bevægelse er Tolmans 16-18 elektronskalregel (afsnit 2).

Reaktioner af koordinerede ligander
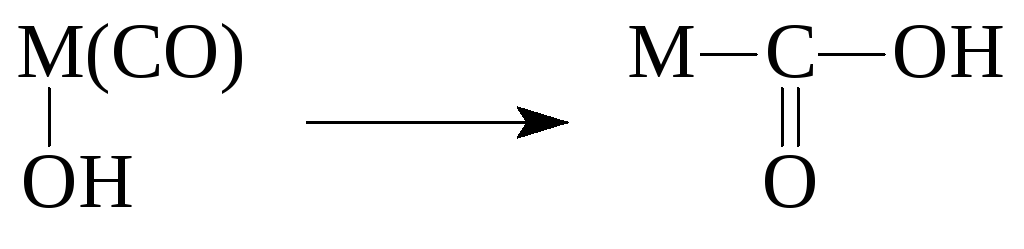








Reaktioner af organometalliske forbindelser



Vi råder dig til at læse





